EXHIBIT 99.1
Published on November 13, 2019
Exhibit 99.1
![]()
MANAGEMENT’S DISCUSSION AND ANALYSIS OF THE FINANCIAL SITUATION AND OPERATING RESULTS – FOR THE THREE AND SIX-MONTH PERIODS ENDED SEPTEMBER 30, 2019 AND 2018
Introduction
This management’s discussion and analysis (“MD&A”) is presented in order to provide the reader with an overview of the financial results and changes to the financial position of Acasti Pharma Inc. (referred to in this MD&A as “Acasti”, “the Corporation”, “we”, “us” and “our”) as at September 30, 2019 and for the three and six-month periods then ended. This MD&A explains the material variations in the financial statements of operations, financial position and cash flows of Acasti for the three and six-month periods ended September 30, 2019, and 2018.
Market data and certain industry data and forecasts included in this MD&A were obtained from internal corporation surveys, market research, and publicly available information, reports of governmental agencies and industry publications and surveys. We have relied upon industry publications as our primary sources for third-party industry data and forecasts. Industry surveys, publications and forecasts generally state that the information they contain has been obtained from sources believed to be reliable, but that the accuracy and completeness of that information is not guaranteed. We have not independently verified any of the data from third-party sources or the underlying economic assumptions they made. Similarly, internal surveys, industry forecasts and market research, which we believe to be reliable based upon our management’s knowledge of our industry, have not been independently verified. Our estimates involve risks and uncertainties, including assumptions that may prove not to be accurate, and these estimates and certain industry data are subject to change based on various factors, including those discussed under “Risk Factors” in this MD&A. While we believe our internal business research is reliable and the market definitions we use in this MD&A are appropriate, neither our business research nor the definitions we use have been verified by any independent source. This MD&A may only be used for the purpose for which it has been published.
In this MD&A, financial information is for the three and six-month periods ended September 30, 2019 and 2018 and is based on the interim financial statements of the Corporation, which were prepared in accordance with International Accounting Standard (“IAS”) 34, Interim Financial Reporting, as issued by the International Accounting Standards Board (“IASB”). The Corporation applied the same accounting policies in the preparation of these condensed interim financial statements as those disclosed in note 3 of its most recent annual financial statements, except they include the new standards, and amendments to standards, which are effective for the period beginning on April 1, 2019 and have been adopted by the Corporation. In accordance with its mandate, the Audit Committee of the Corporation’s Board of Directors reviews the contents of the MD&A and recommends its approval to the Board of Directors. The Board of Directors approved this MD&A on November 13, 2019. Disclosure contained in this document is current to that date, unless otherwise noted. Note that there have been no significant changes to the “Use of estimates and measurement uncertainty”, “Critical Accounting Policies”, and “Financial instruments” in comparison to those disclosed in the Corporation’s MD&A for the year ended March 31, 2019, filed with the securities regulatory authorities on June 26, 2019. Readers should carefully review and consider the risks and uncertainties described in the Corporation’s filings with securities regulators, as well as in its Annual Report on Form 20-F filed with securities regulatory authorities in the U.S. on June 26, 2019 and in Canada on July 4, 2019. The Corporation’s financial results are published in Canadian dollars. All amounts disclosed in this MD&A are in thousands of Canadian dollars, except share and per share amounts or unless otherwise indicated.
1
Additional information about the Corporation can be found on the SEDAR website at www.sedar.com or on EDGAR at www.sec.gov/edgar.shtml under Acasti Pharma Inc.
The Class A shares of the Corporation (“Common Shares”) are listed for trading on the TSX Venture Exchange and on the NASDAQ Capital Market exchange under the ticker symbol “ACST”.
We own or have rights to trademarks, service marks or trade names that we use in connection with the operation of our business. In addition, our name, logo and website names and addresses are our service marks or trademarks. CaPre® is our registered trademark. The other trademarks, trade names and service marks appearing in this MD&A are the property of their respective owners. Solely for convenience, the trademarks, service marks, tradenames and copyrights referred to in this MD&A are listed without the ©, ® and TM symbols, but we will assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensors to these trademarks, service marks and tradenames.
FORWARD-LOOKING STATEMENTS
This MD&A contains information that may be forward-looking information within the meaning of Canadian securities laws and forward-looking statements within the meaning of U.S. federal securities laws, both of which we refer to in this MD&A as forward-looking information. Forward-looking information can be identified by the use of terms such as “may”, “will”, “should”, “expect”, “plan”, “anticipate”, “believe”, “intend”, “estimate”, “predict”, “potential”, “continue” or other similar expressions concerning matters that are not statements about the present or historical facts. Forward-looking information in this MD&A includes, among other things, information or statements about:
| · | our ability to conduct all required clinical and nonclinical trials for CaPre, including the timing and results of those trials; |
| · | our strategy, future operations, prospects and the plans of our management; |
| · | the regulatory plan, timeline, costs and results of our clinical and nonclinical trials for CaPre; |
| · | the timing and outcome of our meetings and discussions with the U.S. Food and Drug Administration, or FDA; |
| · | our planned regulatory filings for CaPre, and their timing; |
| · | our expectation that our Bridging Study (as defined below) results will support our plan to get authorization from the FDA to use the 505(b)(2) pathway with new chemical entity, or NCE, status towards a New Drug Application, or NDA, approval in the United States; |
| · | the timing and results from the STRENGTH study (conducted by Astra Zeneca with their omega-3 (OM3) drug EPANOVA) in patients with high triglycerides, or TGs (blood levels between 200-499 mg/dL) and concomittantly taking a statin; |
| · | the potential benefits and risks of CaPre as compared to other products in the pharmaceutical, medical food and natural health products markets; |
| · | our estimates of the size of the potential market for CaPre, unmet medical needs in that market, the potential for market expansion, and the rate and degree of market acceptance of CaPre if it reaches commercialization, and our ability to serve that market; |
| · | our anticipated marketing advantages and product differentiation of CaPre and its potential to become a best-in-class OM3 compound for the treatment of HTG; |
| · | the potential to expand CaPre’s indication for the treatment of high TGs (200-499 mg/dL); |
| · | the degree to which physicians would switch their patients to a product with CaPre’s target product profile; |
| · | our strategy and ability to develop, commercialize and distribute CaPre in the United States and elsewhere; |
| · | the manufacturing scale-up of CaPre beyond 20 tons per year and the related timing; |
| · | our ability to strengthen our patent portfolio and other means of protecting our intellectual property rights, including our ability to obtain additional patent protection for CaPre; |
2
| · | our expectation that following expiration of the license agreement with Neptune Wellness Solutions Inc. (formerly Neptune Technologies & Bioressources Inc.) (“Neptune”) we will not require any license from third parties to support the commercialization of CaPre; |
| · | the availability, consistency and sources of our raw materials, including krill oil; |
| · | our expectation to be able to rely on third parties to manufacture CaPre whose manufacturing processes and facilities are in compliance with current good manufacturing practices, or cGMP; |
| · | the potential for Omega-3 therapeutics, or OM3s in other cardiometabolic medicine indications; |
| · | our intention and ability to build a U.S. commercial organization and to successfully launch CaPre and compete in the U.S. market; |
| · | our intention and ability to complete development and/or distribution partnerships to support the commercialization of CaPre outside of the United States, and to pursue strategic opportunities to provide capital and market access; |
| · | our need for additional financing and our estimates regarding our future financing and capital requirements; |
| · | our expectation regarding our financial performance, including our revenues, profitability, research and development, costs and expenses, gross margins, liquidity, capital resources, and capital expenditures; and |
| · | our projected capital requirements to fund our anticipated expenses, including our research and development and general and administrative expenses, and capital expenditures. |
Although the forward-looking information in this MD&A is based upon what we believe are reasonable assumptions, you should not place undue reliance on that forward-looking information since actual results may vary materially from it. Important assumptions made by us when making forward-looking statements include, among other things, assumptions by us that:
| · | we are able to obtain the additional capital and financing we require; |
| · | we successfully and timely complete all required clinical and nonclinical trials necessary for regulatory approval of CaPre; |
| · | the timeline and costs for our clinical and nonclinical programs are not materially underestimated or affected by unforeseen circumstances; |
| · | CaPre is safe and effective; |
| · | FDA will grant Amarin an expanded label for CVD risk reduction in patients with mild to moderate HTG |
| · | outcome data from the STRENGTH study is positive; |
| · | we obtain and maintain regulatory approval for CaPre on a timely basis; |
| · | we are able to attract, hire and retain key management and skilled scientific and commercial personnel; |
| · | third parties provide their services to us on a timely and effective basis; |
| · | we are able to maintain our required supply of raw materials, including krill oil; |
| · | we are able to scale-up production of CaPre with third-party manufacturers to support commercial demand; |
| · | we are able to successfully build a commercial organization, launch CaPre in the United States, and compete in the United States market; |
| · | we are able to secure distribution arrangements for CaPre outside of the US, if it reaches commercialization; |
| · | we are able to manage our future growth effectively; |
| · | we are able to gain acceptance of CaPre in its markets and we are able to serve those markets; |
| · | our patent portfolio is sufficient and valid; |
| · | we are able to secure and defend our intellectual property rights and to avoid infringing upon the intellectual property rights of third parties; |
3
| · | we are able to take advantage of business opportunities in the pharmaceutical industry; |
| · | we are able to execute on strategic partnerships according to our business plan; |
| · | we are able to continue as a going concern; |
| · | there is no significant increase in competition for CaPre from other companies in the pharmaceutical, medical food and natural health product industries; |
| · | CaPre would be viewed favorably by payers at launch and receive appropriate healthcare reimbursement; |
| · | market data and reports reviewed by us are accurate; |
| · | there are no adverse changes in relevant laws or regulations; and |
| · | we face no product liability lawsuits and other proceedings or any such matters, if they arise, are satisfactorily resolved. |
In addition, the forward-looking information in this MD&A is subject to a number of known and unknown risks, uncertainties and other factors, including those described in this MD&A under the heading “Risk Factors”, many of which are beyond our control, that could cause our actual results and developments to differ materially from those that are disclosed in or implied by the forward-looking information, including, among others:
| · | risks related to timing and possible difficulties, delays or failures in our ongoing TRILOGY Phase 3 program for CaPre; |
| · | nonclinical and clinical trials may be more costly or take longer to complete than anticipated, and may never be completed, or may not generate results that warrant future development or commercialization of CaPre; |
| · | CaPre may not prove to be as safe and effective or as potent as we currently believe; |
| · | our planned TRILOGY Phase 3 program may not achieve all or any of its primary and secondary endpoints; |
| · | our anticipated studies and submissions to the FDA may not occur as currently anticipated, or at all; |
| · | the FDA could reject our 505(b)(2) regulatory pathway and/or our NDA; |
| · | while the REDUCE-IT results (a Cardiovascular outcome study conducted by Amarin with their OM3 drug VASCEPA) were positive, the cardiovascular outcome study data from the STRENGTH study could be negative, which could also negatively affect the market perception of the class and CaPre; |
| · | we may encounter difficulties, delays or failures in obtaining regulatory approvals for the initiation of clinical trials or to market CaPre, or the FDA may refuse to approve CaPre, or place restrictions on our ability to commercialize CaPre; |
| · | the FDA may require, or for competitive reasons we may need to conduct additional future clinical trials for CaPre, the occurrence and success of which cannot be assured; |
| · | CaPre may have unknown side effects; |
| · | the FDA may refuse to approve CaPre, or place restrictions on our ability to commercialize CaPre; |
| · | CaPre could be subject to extensive post-market obligations and continued regulatory review, which may result in significant additional expense and affect sales, marketing and profitability; |
| · | we may fail to achieve our publicly announced milestones on time; |
| · | we may encounter difficulties in completing the development and commercialization of CaPre; |
| · | third parties we will rely upon to conduct our TRILOGY Phase 3 program for CaPre may not effectively fulfill their obligations to us, including complying with FDA requirements; |
| · | there may be difficulties, delays, or failures in obtaining health care reimbursements for CaPre; |
| · | recently enacted and future laws may increase the difficulty and cost for us to obtain marketing approval of and commercialize CaPre and affect the prices we can charge; |
4
| · | new laws, regulatory requirements, and the continuing efforts of governmental and third-party payors to contain or reduce the costs of healthcare through various means could adversely affect our business; |
| · | the market opportunity for, and demand and market acceptance of, CaPre may not be as strong as we anticipate; |
| · | third parties that we will rely upon to manufacture, supply and distribute CaPre may not effectively fulfill their obligations to us, including complying with FDA requirements; |
| · | there may not be an adequate supply of raw materials, including krill oil, in sufficient quantities and quality and to produce CaPre under cGMP standards; |
| · | we may not be able to meet applicable regulatory standards for the manufacture of CaPre or scale-up our manufacturing successfully; |
| · | as a development stage corporation, we have limited sales, marketing and distribution personnel and resources; |
| · | our patent applications may not result in issued patents, our issued patents may be circumvented or challenged and ultimately struck down, and we may not be able to successfully protect our trade secrets or other confidential proprietary information; |
| · | we may face claims of infringement of third party intellectual property and other proprietary rights; |
| · | we may face product liability claims and product recalls; |
| · | we may face intense competition from other companies in the pharmaceutical, medical food and natural health product industries; |
| · | we have a history of negative operating cash flow and may never become profitable or be able to sustain profitability; |
| · | we have significant additional future capital needs and may not be able to raise additional financing required to fund further research and development, clinical studies, obtain regulatory approvals, build a commercial organization in the United States, and meet ongoing capital requirements to continue our current operations on commercially acceptable terms or at all; |
| · | we may not be able to successfully compete in the United States market with competitors who are larger and have more resources than we do; |
| · | we may acquire businesses or products or form strategic partnerships in the future that may not be successful; |
| · | we may be unable to secure development and/or distribution partnerships to support the development and commercialization of CaPre outside the United States, provide development capital, or market access; |
| · | we rely on the retention of key management and skilled scientific, manufacturing, regulatory and commercial personnel; and |
| · | general changes in economic and capital market conditions could adversely affect us. |
All of the forward-looking information in this MD&A is qualified by this cautionary statement. There can be no guarantee that the results or developments that we anticipate will be realized or, even if substantially realized, that they will have the consequences or effects on our business, financial condition or results of operations that we anticipate. As a result, you should not place undue reliance on the forward-looking information. Except as required by applicable law, we do not undertake to update or amend any forward-looking information, whether as a result of new information, future events or otherwise. All forward-looking information is made as of the date of this MD&A.
5
Caution Regarding Non-IFRS Financial Measures
The Corporation uses multiple financial measures for the review of its operating performance. These measures are generally IFRS financial measures, but one adjusted financial measure, Non-IFRS operating loss, is also used to assess its operating performance. This non-IFRS financial measure is directly derived from the Corporation’s financial statements and is presented in a consistent manner. The Corporation uses this measure, in addition to the IFRS financial measures, for the purposes of evaluating its historical and prospective financial performance, as well as its performance relative to competitors and to plan and forecast future periods as well as to make operational and strategic decisions. The Corporation believes that providing this Non-IFRS information to investors, in addition to IFRS measures, allows them to see the Corporation’s results through the eyes of management, and to better understand its historical and future financial performance.
Earnings and other measures adjusted to a basis other than IFRS do not have standardized meanings and are unlikely to be comparable to similar measures used by other companies. Accordingly, they should not be considered in isolation. The Corporation uses Non-IFRS operating loss to measure its performance from one period to the next without the variation caused by certain adjustments that could potentially distort the analysis of trends in its operating performance, and because the Corporation believes it provides meaningful information on the Corporation’s financial condition and operating results. Acasti’s method for calculating Non-IFRS operating loss may differ from that used by other corporations.
Acasti calculates its Non-IFRS operating loss measurement by adding to net loss its finance expenses, which includes change in fair value of derivative warrant liabilities and foreign exchange gain (loss), depreciation and amortization, impairment loss, litigation settlement that was settled via the issuance of common shares, and stock-based compensation and by subtracting finance income and deferred tax recovery. Items that do not impact core operating performance of the Corporation are excluded from the calculation as they may vary significantly from one period to another. Acasti also excludes the effects of certain non-monetary transactions recorded, such as stock-based compensation and litigation settlement that was settled via the issuance common shares, from its Non-IFRS operating loss calculation. Excluding this item does not imply it is necessarily non-recurring.
A reconciliation of net loss to Non-IFRS operating loss is presented later in this MD&A.
6
BUSINESS OVERVIEW
Our Business
We are a biopharmaceutical innovator focused on the research, development and commercialization of prescription drugs using omega-3 fatty acids, or OM3, delivered both as free fatty acids and bound-to-phospholipid esters, or PLs, derived from krill oil. OM3 fatty acids have extensive clinical evidence of safety and efficacy in lowering triglycerides, or TGs, in patients with hypertriglyceridemia, or HTG. Our lead product candidate is CaPre, an OM3 phospholipid therapeutic, which we are developing initially for the treatment of severe HTG, a condition characterized by very high or severe levels of TGs in the bloodstream (≥ 500 mg/dL). In accordance with a study published in 2009 in the Archives of Internal Medicine by Ford et al., it is estimated that three to four million people in the United States have severe HTG. In primary qualitative market research studies commissioned by Acasti in August 2016 and November of 2017 by DP Analytics, a division of Destum Partners, Key Opinion Leaders (KOLs), High Volume Prescribers (HVPs) and Pharmacy Benefit Managers who were interviewed indicated a significant unmet medical need exists for an effective, safe and well-absorbing OM3 therapeutic that can also demonstrate a positive impact on the major blood lipids associated with cardiovascular disease, or CVD, risk. We believe that CaPre will address this unmet medical need, if our Phase 3 results reproduce what we observed in our Phase 2 data. We initiated TRILOGY, our Phase 3 clinical program in North America, during the second half of 2017, and started clinical site activation as planned at the end of 2017. As of the date of this quarterly report, patient enrollment and randomization have been completed, and the two TRILOGY Phase 3 studies continue on schedule to report topline results by December 2019 for TRILOGY 1, and January 2020 for TRILOGY 2. We also believe the potential exists to expand CaPre’s initial indication to the roughly 36 million patients with high TGs in the mild to moderate range (e.g., blood levels between 200 - 499 mg/dL), although at least one additional clinical trial would likely be required to support FDA approval of a Supplemental New Drug Application (SNDA) to expand CaPre’s indication to this segment. Data from our Phase 2 studies indicated that CaPre may have a positive effect in diabetes and other inflammatory and cardiometabolic diseases; consequently, we may also seek to identify new potential indications for CaPre that may be appropriate for future studies and pipeline expansion. In addition, we may also seek to in-license other cardiometabolic drug candidates for drug development and commercialization.
In four clinical trials conducted to date, we saw the following consistent results with CaPre, and we are seeking to demonstrate similar safety and efficacy in our TRILOGY Phase 3 program:
| · |
significant reduction of TGs and non-high density lipoprotein cholesterol (non-HDL-C) levels in the blood of patients with mild to severe HTG;
|
|
| · |
no deleterious effect on low-density lipoprotein cholesterol (LDL-C), or “bad” cholesterol, with the potential to reduce LDL-C;
|
|
| · |
potential to increase high-density lipoprotein cholesterol (HDL-C), or “good” cholesterol;
|
|
| · |
potential to benefit diabetes patients by decreasing hemoglobin A1c (HbA1c), a marker of glucose control;
|
|
| · |
good bioavailability (absorption by the body), even under fasting conditions;
|
|
| · |
no significant food effect when taken with either low-fat or high-fat meals; and
|
|
| · | an overall safety profile similar to that demonstrated by currently marketed OM3s. |
We believe that if we are able to reproduce these results in our TRILOGY Phase 3 program, that this could potentially set CaPre apart from current FDA-approved fish oil-derived OM3 treatment options, and it could give us a significant clinical and marketing advantage.
7
About Hypertriglyceridemia (HTG)
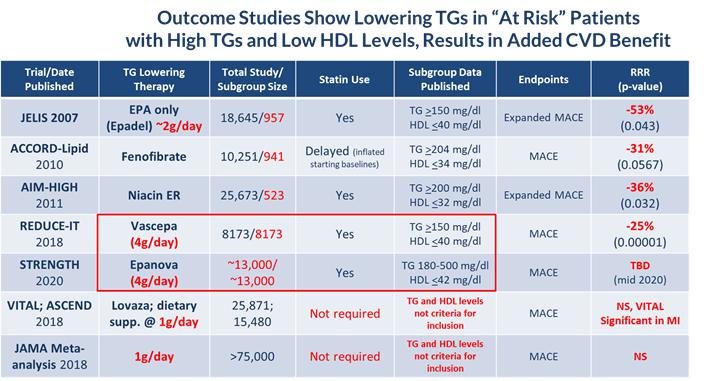
According to the American Heart Association Scientific Statement on Triglycerides and Cardiovascular Disease from 2011, TG levels provide important information as a marker associated with the risk for heart disease and stroke, especially when an individual also has low levels of HDL-C and elevated levels of LDL-C. HTG can be caused by both genetic and environmental factors, including obesity, sedentary lifestyle and high-fat diets. HTG is also associated with comorbid conditions such as chronic renal failure, pancreatitis, nephrotic syndrome, and diabetes. Multiple epidemiological, clinical, genetic studies suggest that patients with elevated TG levels (≥ 200 mg/dL) are at a greater risk of coronary artery disease, or CAD, and pancreatitis, a life-threatening condition, as compared to those with normal TG levels. The genes regulating TGs and LDL-C are equally strong predictors of CAD. Other studies suggest that lowering and managing TG levels may reduce these risks. In addition, the Japan EPA Lipid Intervention Study, or JELIS, demonstrated the long-term benefit of an OM3 eicosapentaenoic acid, or EPA, in preventing major coronary events in hypercholesterolemic patients receiving statin treatment. JELIS found a 19% relative risk reduction in major coronary events in patients with relatively normal TGs but a more pronounced 53% reduction in the subgroup of patients with TGs > 150mg/dL and HDL-C < 40mg/dL. Recently published meta-analyses by Alexander et al. (Mayo Clinic Proceedings, 2017) and Maki et al. (Journal of Clinical Lipidology, 2016) suggest that EPA and docosahexaenoic acid, or DHA, may be associated with reducing coronary heart disease risk to a greater extent in populations with elevated TG levels, and that drugs lowering TG and TG-rich lipoproteins may reduce cardiovascular event risk in patients with elevated TG levels, particularly if associated with low HDL-C. More recently in November of 2018, Amarin published the results of their REDUCE-IT cardiovascular outcome trial (CVOT), which showed that a therapeutic dose of VASCEPA at 4 grams per day, taken on top of a statin, reduced residual cardiovascular risk by 25%. Based on this data, Amarin is currently seeking an expanded label for VASCEPA that would allow use in patients with mild to moderate HTG (200 – 500mg/dl). Astra Zeneca is currently investigating the potential for EPANOVA, their therapeutic OM3 containing both EPA and DHA, taken with a statin to reduce cardiovascular risk in patients with elevated levels of TGs and low HDL-C in their ongoing STRENGTH CVOT, the results of which are expected to be reported sometime in the second half of 2020. The table below lists several CVOT studies done over the last 10 years or so, and supports the current thinking that the right dose of any drug (e.g. OM3, Fibrate or Niacin) that reduces TG levels in at risk patients (e.g. those with elevated TGs and low HDL-C), can significantly reduce their cardiovascular risk.
8
About CaPre
CaPre is a highly purified, proprietary krill oil-derived mixture containing polyunsaturated fatty acids, or PUFAs, primarily composed of OM3 fatty acids, principally eicosapentaenoic acid, or EPA, and docosahexaenoic acid, or DHA, present as a combination of phospholipid esters and free fatty acids. EPA and DHA are well known to be complementary and beneficial for human health, and according to numerous recent clinical studies, may promote healthy heart, brain and visual function (Kwantes and Grundmann, Journal of Dietary Supplements, 2014), and may also contribute to reducing inflammation and blood levels of TGs (Ulven and Holven, Vascular health and risk management, 2015). Krill is a rich natural source of phospholipids and OM3 fatty acids. The EPA and DHA contained in CaPre are delivered as a combination of OM3s as free fatty acids and OM3s bound to phospholipid esters. Both forms allow these PUFAs to reach the small intestine where they undergo rapid absorption and transformation into complex fat molecules that are required for lipid transport in the bloodstream. We believe that EPA and DHA are more efficiently transported by phospholipids sourced from krill oil than the EPA and DHA contained in fish oil, which are transported either by TGs (as in dietary supplements) or as ethyl esters as in other prescription OM3 drugs (such as LOVAZA and VASCEPA). These OM3 ethyl ester prescription products must undergo additional digestion before they are ready for transport into the bloodstream. The digestion and absorption of OM3 ethyl ester drugs requires a particular enzymatic process that is highly dependent on the fat content of a meal – the higher the fat content, the better the OM3 ethyl ester absorption. High fat content meals are not recommended in patients with HTG. We believe that CaPre’s superior absorption profile could represent a significant clinical advantage, since taking it with a low-fat meal represents a healthier and more realistic regimen for patients with HTG who must follow a restricted low-fat diet. CaPre is intended to be used as a therapy combined with positive lifestyle changes, such as a healthy diet and exercise, and can be administered either alone or with other drug treatment regimens such as fibrates and/or statins (a class of drug used to reduce LDL-C). CaPre is intended to be taken orally once or twice per day in capsule form.
Potential Market for CaPre
We believe a significant opportunity exists for OM3 market expansion because, among other things:
| · | Cardiovascular diseases, or CVD, and stroke are the leading causes of morbidity and mortality in the United States. The burden of CVD and stroke in terms of life-years lost, diminished quality of life, and direct and indirect medical costs also remains enormous. According to the American Heart Association, in 2016, CVD cost the American healthcare system $555 billion. By 2035, the cost is estimated to increase to $1.1 trillion; |
| · | Evidence suggests the potential for OM3s as an adjunct to standard treatment in other cardiometabolic indications, such as diabetes and high blood pressure; |
| · | Subgroup analyses from outcome studies conducted since 2007 such as JELIS, ACCORD-Lipid and AIM-HIGH, have all shown that patients who entered these studies with high TGs (above 150 mg/dl) and low HDL (below 40 mg/dl) and received a TG-lowering medication (either an OM3, fibrate or niacin) saw a relative cardiovascular risk reduction of 31 – 53% by the end of the study when compared to placebo or standard of care; |
| · | Based on the assumption that the REDUCE-IT trial sponsored by Amarin and the STRENGTH trial sponsored by Astra Zeneca, would be positive, key opinion leaders interviewed by DP Analytics in the market research study conducted in 2018 before the results of REDUCE-IT were announced and described further below, estimated that they would increase their own prescribing of OM3s by 43% in patients with high TGs (blood levels between 200 – 499 mg/dL) and by 35% in patients with severe HTG (based on qualitative market research with Key Opinion Leaders (KOLs) and High Volume Prescribers (HVPs) conducted for Acasti in November, 2017 by Destum Partners, an independent market research firm); |
| · | In February 2019, following the release of the REDUCE-IT results in September 2018, Cantor Fitzgerald projected that based on their market research, prescriptions for OM3s are expected to grow in 2019 by 100%. The most recent (March 2019) audited prescription data from Symphony Health Analytics indicates that VASCEPA sales in August, 2019 had increased by 101% over August, 2018; and |
| · | Several analysts who cover the HTG segment of the market are now projecting that this market could reach $5 to $10 billion or more in the US alone over the next few years. |
9
According to the American Heart Association, the prevalence of HTG in the United States and globally correlates to the aging of the population and the increasing incidence of obesity and diabetes. Market participants, including the American Heart Association, have estimated that one-third of adults (approximately 70 million people) in the United States have elevated levels of TGs (TGs >150 mg/dL) (Ford, Archives of Internal Medicine, 2009; 169(6):572-578), including approximately 3 to 4 million people diagnosed with severe HTG (Miller et al. Circulation, 2011 and Maki et al. J. Clan. Lipid, 2012). Moreover, according to Ford, Archives of Internal Medicine in a study conducted between 1999 and 2004, 18% of adults in the United States, corresponding to approximately 40 million people, had elevated TG levels equal to or greater than 200 mg/dl, of which only 3.6% were treated specifically with TG-lowering medication (Ford, Archives of Internal Medicine, 2009; 169(6):572-578; Kapoor and Miller, ACC, 2016, Christian et al. Am. J. Cardiology, 2011). We believe this data indicates there is a large underserved market opportunity for CaPre.
CaPre’s target market in the United States for treatment of HTG was estimated by Symphony Health Analytics Audit data to be approximately US$1.4 billion in 2018, with approximately 4.5 million prescriptions written annually. The total global market for treatment of HTG was estimated by GOED Proprietary Research in 2015 to be approximately US$2.3 billion annually. Currently, all marketed OM3 products are approved by the FDA only for patients with severe HTG. We believe there is the potential to greatly expand the treatable market in the United States to the approximately 70 million people with TGs above 135 mg/dl, assuming the FDA approves expanded labeling for VASCEPA based on the recent positive REDUCE-IT outcome study results, and favorable results are reported from the STRENGTH outcome trial, which is currently ongoing and expected to report sometime in 2020. These CVOT studies were designed to evaluate the long-term benefit of lowering TGs on CVD risk with prescription drugs containing OM3 fatty acids in patients with mild to moderately elevated TGs, low HDL-C, and concurrently taking a statin. Additional clinical trials would likely be required for CaPre to also expand its label claims to this segment. Given the large portion of the adult population in the United States that have elevated levels of TGs above 135 mg/dL but who go largely untreated, we believe there is the potential for a very significant increase in the total number of patients eligible for treatment based on the positive REDUCE-IT results and provided the outcome of the STRENGTH trial is also positive.
CaPre currently has two FDA-approved and marketed branded competitors (LOVAZA and VASCEPA). In addition, Astra Zeneca has an FDA-approved product, EPANOVA, which has not yet been launched. Generic LOVAZA became available on the U.S. market in 2013. In spite of generic options, 2017 audited prescription data from IMS NSP indicates that approximately 70% of OM3 prescriptions were written for branded products (predominantly VASCEPA). According to the most recently available Symphony Health Analytics Audit data from August 2019, the U.S. OM3 market for HTG is valued at more than $1.6 billion. However, the number of prescriptions written for branded OM3s is now increasing significantly since Amarin announced its REDUCE-IT results in late 2018. Several analysts are predicting that this trend will continue, driving substantial market growth.
We conduct market research at least annually with physicians and payers to monitor market developments, reimbursement and clinical practice. Except as otherwise indicated, all of the information that follows under this section has been derived from secondary sources, including audited U.S. prescribing data, and from qualitative U.S. primary market research with physicians and payers conducted for us by DP Analytics, a division of Destum Partners, Inc., or Destum, and more recently by Medical Marketing Economics (MME).
Destum utilized secondary market data and reports to develop market projections for us, and they also conducted primary qualitative market research with physicians and third-party payers, such as PBMs. One-on-one in-depth phone interviews conducted in November 2017 lasting on average 30-60 minutes were conducted with 22 physicians and 5 PBMs. Key insights and data were collected by Destum on current clinical practice for treating patients with HTG, and physician and payer perceptions of the current unmet medical and key economic needs in this space. All interviews were conducted by the same individual at Destum to ensure consistency in the collection of key information. Destum utilized OM3 prescription data from 2009 to 2017 to estimate the size of CaPre’s potential market. Based on discussions with the PBMs, Destum also assumed CaPre would be viewed favorably by payers at launch (e.g., Tier 2 or 3, depending on payer plan, which is comparable to LOVAZA and VASCEPA), provided CaPre is similarly priced. Upon completing the screening questionnaire and being approved for inclusion in Destum’s study, key opinion leaders (KOLs) and high volume prescribers (HVPs), were provided with a study questionnaire and were asked to comment on a target profile for a potential new OM3 “Product X” delivering a “trifecta” of cardio-metabolic benefits similar to the potential efficacy and safety benefits demonstrated by CaPre in our two Phase 1 pharmacokinetic studies and two Phase 2 clinical trials, which we refer to as the Target Product Profile. Respondents were told that the unidentified product was being prepared for a Phase 3 program designed to confirm with statistical significance the product’s safety and efficacy in patients with severe HTG. The Target Product Profile was used by Destum strictly for market research analysis purposes and should not be construed as an indication of future performance of CaPre and should not be read as an expectation or guarantee of future performance or results of CaPre, and will not necessarily be an accurate indication of whether or not such results will be achieved by CaPre in our Phase 3 program.
10
In the market research for us, KOLs and HVPs interviewed by Destum were asked to assess the level of unmet medical need associated with treating patients with severe HTG based on currently available treatment options. 91% of physicians interviewed by Destum in 2016 indicated that they believe that the current unmet medical need for treating HTG was moderate to high. That number increased to 100% in the subsequent December 2017 research. The reasons identified by these physicians for their dissatisfaction with the currently available OM3s included insufficient lowering of TGs (a complaint principally related to VASCEPA), negative LDL-C effects (a complaint principally related to LOVAZA), the “food effect” or reduced absorption of both LOVAZA and VASCEPA when taken with a low-fat meal (or the corollary to this concern which is that their patients had to take either drug with a fatty meal to get full efficacy benefit), gastrointestinal side effects, and the fishy taste from these fish oil-derived OM3s. Physicians reported that their patients have difficulty swallowing the large 1 gram softgel capsules of VASCEPA and LOVAZA, and they worried about these issues contributing to patient non-compliance. Despite the availability of other drug classes to treat severe HTG, interviewed physicians indicated that they would welcome the introduction of new and improved OM3 products, particularly if they can address these perceived deficiencies.
Interviewed physicians responded favorably to the blinded Target Product Profile of CaPre in the Destum Market Research studies. In the most recent study conducted in December 2017, they indicated that they would prescribe a new OM3 drug with the Target Product Profile to approximately 82% of their patients in the severe HTG patient population and 68% of their patients in the high HTG segment within two years of the new OM3 drug’s approval. Approximately 60% of the interviewed physicians indicated that they would switch to a drug with the Target Product Profile primarily due to the “trifecta effect” of reducing TGs and LDL-C while elevating HDL-C, and the remaining 40% indicated they would switch primarily due to a drug with the Target Product Profile due to the effective reduction of TGs alone. In connection with their responses, the interviewed physicians were instructed to assume the drug with the Target Product Profile and all currently available OM3 products were priced similarly and not subject to any reimbursement or coverage hurdles (e.g., all products were on an equal health care coverage playing field). This assumption was subsequently supported by our interviews with leading PBMs in the United States.
This market research was updated in March 2019 to reflect the current views of physicians and third party payers following the publication of the REDUCE-IT study results. This updated primary qualitative market research project was conducted by Medical Marketing Economics, and the design of the study was similar to the Destum project, with one-on-one interviews lasting approximately 60 minutes in duration. These interviews were conducted with 10 physicians and 20 pharmacy directors, covering 179,913,005 commercial lives across the United States, consistent with the current payer mix for the OM3 market. CaPre was evaluated positively by physicians with particular value placed on its potential to lower TGs, LDL-C, and HbA1c (this was seen as unique, and especially valued), and to increase HDL-C, as well as its potentially superior tolerability features (e.g. easier to swallow when compared to the ethyl ester fish oils, and no fishy taste or "burpiness"). Importantly, since this research was conducted after the REDUCE-IT trial outcome results, the lack of clinical outcomes data for CaPre at launch was generally not seen as problematic for the majority of the physicians interviewed. On average, physicians indicated that they would begin prescribing CaPre 3 months after launch and would evaluate its performance in their initial patients after 3 to 6 months of use. Depending on favorable experience in initial use, some physicians indicated peak use could begin as quickly as 12 to 18 months after launch. Physicians expect CaPre to be priced similar to VASCEPA, and to have an out-of-pocket cost of approximately $10-$50. Payers also viewed CaPre favorably and did not anticipate any major reimbursement restrictions, with likely coverage at Tier 2 or 3 depending on the payer plan.
Based on both primary market research with pharmacy benefit managers, or PBMs, and audited prescription reports, the pricing for branded products currently averages between US$299 and US$355 per month. Amarin has raised prices for VASCEPA annually since its launch in late 2013. PBMs offer “Preferred Brand” status (Tier 2 or Tier 3), without significant restrictions (i.e., no prior authorization, step edits, or high co-payments) for these branded OM3s. By the end of 2018, VASCEPA had reached about 45% market share in the United States, in spite of generic competition from LOVAZA. Amarin continues to gain market share in the United States and, as of August 2019, had reached more than 50% of the market share based on units. This growth is principally coming from market expansion along with some erosion of generic sales.
We plan to continue to regularly conduct additional market research with KOLs, HVPs, primary care physicians and payers to further develop and refine our understanding of the potential market for CaPre ahead of commercial launch in the United States.
11
Our Clinical Data
CaPre is being developed by us for the treatment of patients with severe HTG. In two Phase 2 clinical trials conducted by us in Canada (our COLT and TRIFECTA trials), CaPre was well-tolerated at all doses tested, with no serious adverse events that were considered treatment-related. Among the reported adverse events with an occurrence of greater than 2% of subjects and greater than placebo, only diarrhea had an incidence of 2.2%.
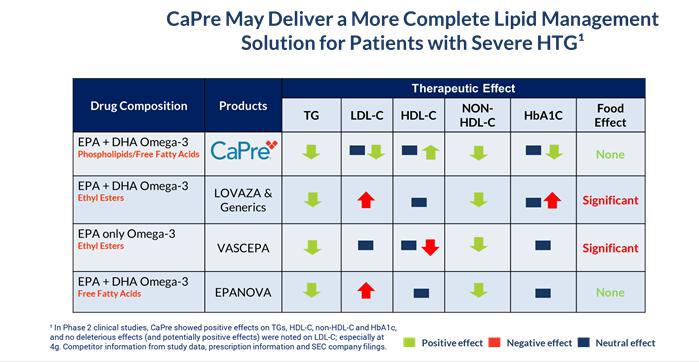
In both Phase 2 clinical trials, CaPre significantly lowered TGs in patients with mild to severe HTG. Importantly, in these studies, CaPre also demonstrated no deleterious effect on LDL-C (unlike LOVAZA and EPANOVA, which had been shown to significantly increase LDL-C in patients with severe HTG). Further, our Phase 2 data indicated that unlike LOVAZA, CaPre may actually reduce LDL-C with a 4 gram per day dose (a dose equivalent to VASCEPA and LOVAZA). LDL-C is undesirable because it accumulates in the walls of blood vessels, where it can cause blockages (atherosclerosis). Clinically, the phospholipids may potentially not only improve the absorption, distribution, and metabolism of omega-3s, but they may also decrease the synthesis of LDL cholesterol in the liver, impede or block cholesterol absorption, and stimulate lipid secretion from bile. In the Phase 2 trials, CaPre also significantly reduced non-HDL-C (all cholesterol contained in the bloodstream except HDL-C), which is also considered to be a marker of cardiovascular disease. The COLT trial data showed a mean increase of 7.7% in HDL-C with CaPre at 4 grams per day (p=0.07). Further analysis of the data from our on-going TRILOGY Phase 3 program will be required to demonstrate CaPre’s statistical significance with respect to lowering LDL-C and increasing HDL-C. Finally, we saw a statistically significant reduction of HbA1c in the CaPre 4g treatment group in the COLT study after only 8 weeks on drug in a diabetic population of patients with HbA1c levels at or below 7.0% at baseline. This interesting and potentially differentiating effect is being investigated more thoroughly in our TRILOGY Phase 3 program, where a larger proportion of the patients are diabetic, with HbA1c levels up to 9.5%, and they will be followed for 6 months.
We believe that these multiple potential cardiometabolic benefits, if confirmed in our on-going TRILOGY Phase 3 program, could be significant differentiators for CaPre in the marketplace, as no currently approved OM3 drug has shown an ability to positively modulate all four of these important blood lipids (TGs, non-HDL-C, LDL-C and HDL-C) in the treatment of patients with dyslipidemia. We also believe that if supported by additional clinical trials, CaPre has the potential to become the best-in-class OM3 compound for the treatment of mild to moderate HTG.
In summary, in addition to effectively reducing TG levels in patients with mild to severe HTG, clinical data collected by us to date indicates that CaPre may also have:
| · | beneficial clinical effects on other blood lipids, such as HDL-C (good cholesterol) and non-HDL-C; | |
| · | no deleterious effect on, and may potentially reduce, LDL-C (bad cholesterol) levels; | |
| · | potential to benefit diabetes patients by reducing HbA1c, an important marker of diabetes; and | |
| · | absorption capability that, unlike VASCEPA and LOVAZA, is not meaningfully affected by the fat content of a meal consumed prior to drug administration, providing patients with the reassurance that following their physician-recommended low-fat diet will still result in high absorption. |
We believe that these features could set CaPre apart from currently available FDA-approved OM3 treatment options in the marketplace and could give us a significant clinical and marketing advantage.
CaPre’s potential clinical benefits as compared to currently available FDA-approved OM3 treatment options are summarized in the table below and indicate that CaPre may deliver a more complete lipid management solution for patients with severe HTG:
12
Our Nonclinical Research
In addition to our Phase 2 clinical trials, we carried out an extensive nonclinical program to demonstrate the safety of CaPre in a defined set of studies required by the FDA. These studies were carried out by contract research organizations in compliance with Good Laboratory Practices (GLPs) and conducted on various species of animals recommended by the FDA to investigate the long-term effects of CaPre at doses of up to 65 grams of human equivalent dose over 39 weeks. In these studies, hematological, biochemical, coagulation and overall health parameters of CaPre were evaluated and no toxic effects were observed in any of the segments of the studies. Other studies focused on the potential toxic effects of CaPre on vital systems, such as the cardiovascular, respiratory and central nervous system as evaluated by behavioral studies of the various species. These studies showed that CaPre did not have any adverse or toxic effects on any of the vital systems investigated, again up to doses well above the recommended clinical dose of CaPre. To rule out short term toxic effects of CaPre on genes, genomic toxicity studies were undertaken on accepted cellular and animal models. These studies showed no toxic effects of CaPre on any of the genetic markers indicative of potential gene altering toxic effects.
We believe the studies conducted to date indicate that CaPre is well-tolerated and shows no toxic effects on any of the physiological and vital systems of the tested animals or their genes at doses well above CaPre’s anticipated clinical therapeutic dose of 4 grams daily.
In parallel to our TRILOGY Phase 3 program, we are currently completing additional nonclinical studies, including a pre- and postnatal development study in rodents and a 26-week oral carcinogenicity study in transgenic homozygous rasH2 mice. Both study protocols were pre-approved by the FDA by means of Special Protocol Assessment (SPA) through the FDA’s Executive Carcinogenicity Assessment Committee. These nonclinical studies are required to support an NDA filing for CaPre.
13
Our TRILOGY Phase 3 Program
In March 2017, we announced our plans to proceed with our TRILOGY Phase 3 program following our End-of-Phase 2 meeting with the FDA in February 2017. Based on the guidance we received from the FDA, we implemented two pivotal, randomized, placebo-controlled, double-blinded Phase 3 studies to evaluate the safety and efficacy of CaPre in patients with severe HTG. These studies of 26-week duration will evaluate CaPre’s ability to lower TGs from baseline in approximately 500 patients (approximately 250 per study) randomized to either 4 grams daily or placebo. The FDA’s feedback supported our plan to conduct two studies in parallel, potentially reducing the cost and shortening the time to an NDA submission. These studies are being conducted in approximately 150 sites across North America.
The primary endpoint of these studies is to determine the efficacy of CaPre at 4 grams/day compared to placebo in lowering TGs after 12 weeks in severe HTG patients, and to confirm safety by continuing to follow these patients for the full 26 weeks. The study was designed to provide at least 90% statistical power to detect a difference of at least a 20% decrease from baseline in TGs between CaPre and placebo. In addition, the Phase 3 studies will include numerous secondary and exploratory endpoints, which are designed to assess the effect of CaPre on the broader lipid profile and certain metabolic, inflammatory and CVD risk markers.
In November 2017, we announced that Dariush Mozaffarian, M.D., Dr.P.H., agreed to serve as the principal investigator of our Phase 3 clinical program. Dr. Mozaffarian is a cardiologist and epidemiologist serving as the Jean Mayer Professor of Nutrition & Medicine, and the Dean of the Friedman School of Nutrition Science & Policy at Tuft’s University. His widely published research focuses on how diets, such as those rich in OM3s, and lifestyle influence cardiometabolic health and how effective policies can improve health and wellness.
Late in 2017, based on feedback from the FDA, we finalized our Chemistry, Manufacturing, and Controls plans and the clinical trial design that supports our TRILOGY Phase 3 program. In parallel with our Phase 3 clinical trial planning, additional current Good Manufacturing Practices (cGMP) production lots of API (known as NKPL66) and CaPre were manufactured, enabling us to build the CaPre and placebo inventory required to support the activated clinical trial sites and complete patient randomization. In the first calendar quarter of 2018, additional lots of CaPre were manufactured for use in our Phase 3 program. With manufacturing of clinical trial material completed mid-2019, we are now allocating our technical resources to other activities related to the scale-up of manufacturing for the planned commercial launch of CaPre in 2021. Acasti recently entered into a supply agreement with a major manufacturer of raw krill oil (RKO), which we anticipate will secure RKO material sufficient for the first two years of commercial launch.
We initiated our TRILOGY Phase 3 program and began site activation and patient enrollment on schedule at the end of 2017. We are working with a major clinical research organization to manage our TRILOGY Phase 3 program. The TRILOGY studies continued to progress on schedule throughout 2018 and 2019, and as of the date of this quarterly report, they remain on schedule for delivery of topline results for TRILOGY 1 in December 2019, and TRILOGY 2 in January 2020. As of the end of September 2019, our two on-going Phase 3 TRILOGY trials had reached 100% patient randomization at more than 150 clinical sites across the United States, Canada, and Mexico, and 85 of the patients in both trials had completed their 6-month treatment plan.
Our first study, designated as TRILOGY 1, is being conducted exclusively in the United States and is fully randomized with a total of more than 250 patients. The TRILOGY 2 study, which is also fully randomized, also has more than 250 patients, and is being conducted in the United States, Canada and Mexico. We expect to report topline results independently for each study as we receive the results.
14
The following chart illustrates the design and dosing of our TRILOGY Phase 3 program for CaPre.
TRILOGY Phase 3 Clinical Program
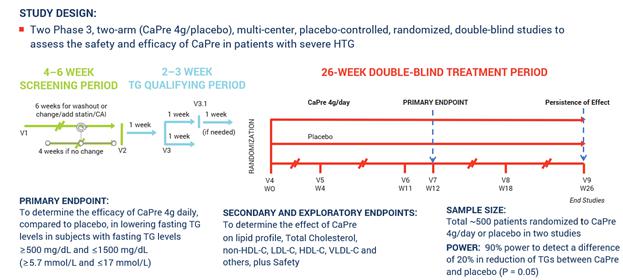
Topline results for TRILOGY will include a readout of the primary endpoint, which is intended to show CaPre’s overall impact on lowering triglycerides (TGs) after 12 weeks compared to placebo. The placebo used in the TRILOGY trials is cornstarch, which is inert, and consequently is expected to have a neutral effect on key biomarkers of patients in the placebo group. As a reminder, the TRILOGY studies are designed to provide at least 90% statistical power to detect a difference of at least a 20% decrease from baseline in TGs between CaPre and placebo. Acasti has shared the statistical analysis plan (SAP) for the analysis and reporting of the TRILOGY results with the FDA, and will finalize it prior to database lock. Subject to any input or changes required by the FDA, Acasti is currently planning that the topline TRILOGY results will include the primary endpoint of TG reduction at Week 12 compared to placebo. Safety and tolerability (e.g. overall adverse events (AE) and serious AE rate, any discontinuation due to AEs, and AEs of special interest such as gastrointestinal events) will also be reported.
The Company currently expects that topline results will not include any secondary or exploratory endpoints. The important secondary and exploratory endpoint results are expected to follow shortly after the release of the topline results of TRILOGY 2, currently anticipated in late January 2020. According to the SAP, the primary endpoint must first be positive with statistical significance prior to analyzing the secondary and exploratory endpoints. These endpoints will then be analyzed in the following order: 1) Additional TG secondary endpoints, including TG reduction at Week 26, which is intended to show CaPre’s persistence of effect, TG reduction in various subgroups to show consistency of effect (such as patients stratified with baseline qualifying TG levels of ≤750 mg/dL vs. >750 mg/dL), and a comparison of TG reduction in patients using and not using statins at baseline, 2) Non-HDL-C, 3) VLDL-C, 4) HDL-C, 5) LDL-C and HbA1c. Physician investigators determined if patients with high LDL-C and/or high HbA1c levels at screening should be put on standard therapy, and if so, they were stabilized prior to being randomized into TRILOGY. Results for both LDL-C and HbA1c will then require subgroup analyses, which are done by combining diabetic patients and separately patients with high LDL-C at baseline from both studies to reach adequate statistical power to detect a difference if one exists, and potentially any incremental benefit of CaPre above the standard of care. Acasti expects that the remaining secondary and exploratory endpoints along with various additional subgroup analyses should be completed before the end of March 2020. In addition to our preliminary topline data, we will seek to present the full data set, which will include results for our key secondary and exploratory endpoints of interest such as LDL-C, VLDL, HDL-C and HbA1c at several scientific meetings in 2020. The Company will communicate more information in the months ahead on how and when all of the TRILOGY results will be reported once the SAP is finalized.
15
Given the positive results we saw from our Phase 2 trials in a total of 675 patients, we eagerly await the completion of the results from our two TRILOGY clinical studies. These trials showed not only a significant reduction of triglycerides, but also indicated that CaPre at a dose of 4g/day may have a positive effect on other major lipid markers such as VLDL, LDL-C, and HDL-C, as well as HbA1c in patients with diabetes. As previously disclosed, the Corporation believes the Phase 3 trial is well designed due to the fact the patients enrolled have higher baseline triglyceride levels (above 500 mg/dl) versus our Phase 2 studies, where most had baseline triglycerides significantly below 500 mg/dl. Additionally, the patients randomized to CaPre in TRILOGY all receive 4 grams per day and will remain on drug for 6 months, while the Phase 2 studies included patients receiving a range of doses from 1 gram, 2 grams and 4 grams per day for only 8 to 12 weeks, which is important given the favorable dose response seen in the Phase 2 studies. Furthermore, based on the information obtained at screening and randomization, it is expected that there will be more patients in the TRILOGY studies on statins, as compared to our Phase 2 studies. It is well known that statins have a synergistic effect on OM3s, resulting in a larger reduction of TGs. Assuming the TRILOGY trials replicate the Phase 2 data, Acasti believes CaPre has the potential to provide an attractive alternative to current therapies, and thus improve the lives of the millions of patients with cardiometabolic disease.
Our Regulatory Strategy for CaPre
Our strategy is to develop and initially commercialize CaPre for the treatment of severe HTG. The TRILOGY Phase 3 program has been designed to evaluate the clinical effect of CaPre on TGs, non-HDL-C, LDL-C, and HDL-C levels together with a variety of other cardiometabolic biomarkers in patients with severe HTG.
We intend to pursue a 505(b)(2) regulatory pathway towards an NDA approval in the United States. A 505(b)(2) regulatory pathway is defined in the U.S. Federal Food Drug and Cosmetic Act (FDCA) as an NDA containing investigations of safety and effectiveness that are being relied upon for approval and were not, in whole, conducted by or for the applicant, and for which the applicant has not obtained a right of reference. 505(b)(2) regulatory pathways differ from a typical NDA because they allow a sponsor to rely, at least in part, on the FDA’s findings of safety and/or effectiveness for a previously approved drug. We intend to pursue the 505(b)(2) regulatory pathway as a strategy to leverage the large body of safety data for LOVAZA, which could accelerate and streamline the development of CaPre and reduce associated costs and risks. We are conducting two Phase 3 studies to independently assess CaPre’s effectiveness in lowering TGs, and its broader effect in patients with cardiometabolic disease. Consequently the use of this 505(b)(2) pathway still allows CaPre to retain its New Chemical Entity (NCE) status due to its novel, patented OM3 free fatty acid/phospholipid ester formulation. In connection with our intended use of the 505(b)(2) pathway, the FDA supported our proposal to conduct our Bridging Study that compared CaPre (which has an OM3 free fatty acid/phospholipid composition) with LOVAZA (which has an OM3-acid ethyl esters composition) in healthy volunteers. In February 2017, we met with the FDA at an End-of-Phase 2 meeting where our Bridging Study data was discussed. We confirmed with the FDA the 505(b)(2) regulatory approach to use the safety data for LOVAZA and finalized the study design for our Phase 3 program that would be required for NDA approval.
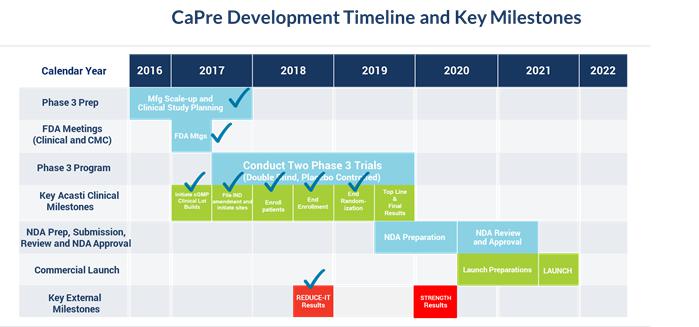
Our planned remaining key development and regulatory milestones and timeline are presented below:
16
Our Intellectual Property Strategy
Under a license agreement we entered into with Neptune in August 2008, which was later amended on February 9, 2009 and March 7, 2013 (the “License Agreement”), we received an exclusive license to use certain intellectual property of Neptune (which includes several patents) to develop and commercialize CaPre and our novel and active pharmaceutical ingredients, or APIs, for use in pharmaceutical and medical food applications in the cardiometabolic field. The term of the License Agreement expires on the date of the last-to-expire licensed patents in 2022. As the result of a royalty prepayment transaction we entered into with Neptune on December 4, 2012, we are not required to pay any royalties to Neptune under the License Agreement during its term for the use of the licensed intellectual property.
Upon the expiry of the License Agreement and related patents, we believe that CaPre will be covered under our own issued and pending patents, and we do not believe that we will afterwards require any licenses to support the commercialization of CaPre.
We currently have patents granted and allowed in the following countries: United States, Canada, Russia, Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, United Kingdom, Italy, Netherlands, Norway, Portugal, Sweden, Japan, Israel, Australia, China, Mexico, Panama, Saudi Arabia, Taiwan, South Africa, Chile and South Korea. We continue to expand our own intellectual property, or IP, patent portfolio. We have filed patent applications in more than 20 jurisdictions, including with the European Patent Office (but excluding the individual countries where we have subsequently registered), and in all major countries in North America, Asia and Australia for our “Concentrated Therapeutic Phospholipid Composition”, or Proprietary Composition, to treat HTG. We currently have 20 issued or allowed patents (including registered European countries) and numerous patent applications pending. A patent is generally valid for 20 years from the date of first filing. However, patent terms can be subject to extensions in some jurisdictions in order to compensate, for example, for delays caused by the patent office during prosecution of the patent application or for regulatory delays during the pre-market approval process.
On January 9, 2019, we announced a Certificate for a European Patent had been issued by the European Patent Office. The granted patent is valid until 2030 and relates to a concentrated phospholipid composition and method of using the same for modulating blood lipids. This patent was validated in Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, United Kingdom, Italy, Netherlands, Norway, Portugal and Sweden. We also received notices of allowances for patents in Chile, Mexico and Israel.
17
We believe these patents and patent applications increase potential commercial opportunities for CaPre, including through possible licensing and partnership opportunities. We are committed to building a global portfolio of patents to ensure long-lasting and comprehensive intellectual property protection and to safeguard potentially valuable market expansion opportunities.
Our patent No. 600167 in New Zealand, which is in force until 2030 and relates to a concentrated phospholipid composition comprising 60% or greater PL concentration and method of using the same for treating cardiovascular diseases, has been opposed by BIO-MER Ltd. The evidentiary stage in the New Zealand patent opposition has been completed. The next step is the hearing. In our view, no new prior art has been presented that was not already considered in other jurisdictions, such as in the United States, where our patents are in force.
We have received a notice issued from the Japan Patent Office (JPO) indicating that a third party filed an opposition against our Japanese Patent No. 6346121. The claim amendment was filed by Acasti and the Company was successful in overcoming the prior art cited in the opposition.
The trademark CaPre® is registered in the United States, Canada, Australia, China, Japan and Europe. We are currently in the process of developing a new brand name and logo for CaPre for launch into the U.S. market. That name, once it is developed, will be trademarked in all of the major jurisdictions around the world. In addition, two PCT applications that cover our encapsulation apparatus and manufacturing process have been filed in all territories under the PCT treaty, while maintaining industrial trade secrets and know-how. We have also filed a provisional application covering our starting material composition (RKO) for the use of CaPre manufacturing.
Our Business and Commercialization Strategy
Key elements of our business and commercialization strategy include initially obtaining regulatory approval for CaPre in the United States for severe HTG. We plan to launch CaPre ourselves in the U.S. market. Our preferred strategy outside the United States is to commercialize CaPre through regional or country-specific strategic partnerships, and to potentially seek support and funding from each partner for in-country clinical development, registration and commercialization activities. We believe that a late development-stage and differentiated drug candidate like CaPre could be attractive to various global, regional or specialty pharmaceutical companies, and we are taking a targeted approach to partnering and licensing in various geographies.
Our key commercialization goals include:
| · | complete our TRILOGY Phase 3 program and, assuming the results are positive, file an NDA by mid-2020 to obtain regulatory approval for CaPre in the United States, initially for the treatment of severe HTG, with the potential to afterwards expand CaPre’s indication to the treatment of high TGs (although at least one additional clinical trial would likely be required to expand CaPre’s indication to this segment), and possibly other additional indications; |
| · | continue to strengthen our patent portfolio and other intellectual property rights; |
| · | continue planning for the potential launch of CaPre in the United States by the second half of 2021; and |
| · | continue to pursue strategic opportunities outside of the United States, such as licensing or similar transactions, joint ventures, partnerships, strategic alliances or alternative financing transactions, to provide development capital, market access and other strategic sources of capital. |
| · | continue to search for attractive assets for in-licensing and/or acquisition that could be synergistic with CaPre, and leverage our commercial organization. |
We expect that additional time and capital will be required to complete the filing of an NDA to obtain FDA approval for CaPre in the United States, and to complete business development collaborations, marketing and other pre-commercialization activities before reaching the commercial launch of CaPre in the United States.
18
Recent Developments
| 1. | On June 24, 2019, Acasti announced that it has received a Notice of Allowance for its second patent in the People’s Republic of China. The new patent is valid until 2030, and expands existing claims related to concentrated therapeutic omega-3 phospholipid compositions and covers certain methods for the treatment and prevention of cardiovascular diseases, metabolic syndrome, inflammation, and neurodevelopmental and neurodegenerative diseases. |
| 2. | On August 9, 2019, Acasti announced that the Company participated on a lipid panel entitled “Lipids: Moving Beyond Statins: Omega 3s, Bempedoic, and More,” moderated by Harold Bays MD, a key opinion leader within the space during the 2019 BTIG Biotechnology Conference. |
| 3. | On August 14, 2019, as part of the Q1 business update, Acasti reported that approximately $8.1 million in proceeds had been received from the recent exercise of warrants since July 1, 2019, which further extends the Company’s cash runway through June of 2020 based on management’s current projections. |
| 4. | On August 28, 2019, Acasti announced the voting results for the matters listed in its management circular dated July 24, 2019 held at its Annual and Special Meeting of Shareholders, in Laval, Canada on August 27, 2019 (the “AGM”). At the AGM, the following individuals were elected as directors of the Corporation for the ensuing year: Roderick N. Carter, Jean-Marie (John) Canan, Jan D’Alvise and Donald Olds. KPMG LLP were also appointed as the Corporation’s auditors for the ensuing fiscal year and the directors were authorized to fix their remuneration. Shareholders also approved various amendments to the Corporation Stock Option Plan and Equity Incentive Plan. |
| 5. | On September 9, 2019, Acasti announced that the Company was awarded up to $750,000 in non-dilutive and non-repayable funding, as well as technical and business advisory services, from the National Research Council of Canada Industrial Research Assistance Program (NRC IRAP) to apply towards eligible research and development disbursements of the Company’s unique commercial production platform for CaPre. With NRC IRAP support, Acasti aims to further expand and enhance its production capabilities for CaPre. |
| 6. | On September 30, 2019, Acasti announced that 100% of the required total patients for our two Phase 3 studies had been randomized, and nearly 80% of the patients in both studies combined had completed their 6-month plans. This progress further supports management’s confidence in announcing topline results of TRILOGY 1 before the end of calendar 2019 and topline results of TRILOGY 2 in January 2020. |
| 7. | On September 30, 2019, Acasti made the determination that the Company will migrate from reporting in IFRS to US GAAP effective beginning with Q4, FY’20 (March 31, 2020 year-end) reports. |
| 8. | On November 4, 2019, Acasti announced that it had partnered with Aker BioMarine to deliver raw krill oil (RKO) to Acasti, under a two-year, fixed price supply agreement. |
| 9. | On November 7, 2019, Acasti announced the publication of a CaPre pharmacokinetics study entitled, “Evaluation of OM3-PL/FFA Pharmacokinetics After Single and Multiple Oral Doses in Healthy Volunteers” in a leading peer-reviewed journal, Clinical Therapeutics. The study showed that the bioavailability of CaPre did not appear to be meaningfully affected by the fat content of the meal consumed before dose administration. |
19
Basis of presentation of the financial statements
The Corporation is subject to a number of risks associated with its ongoing priorities, including the conduct of its clinical program and its results, the establishment of strategic alliances and the development of new pharmaceutical products and their marketing. The Corporation’s current product in development requires approval from the U.S Food and Drug Administration and equivalent regulatory organizations in other countries before sale can be authorized. Certain risks have been reduced for the longer term with the outcome of the Corporation’s actions, including its intellectual property strategy execution with filed patent applications in more than 20 jurisdictions, with 20 issued patents and with numerous additional patent applications pending. The Corporation has incurred significant operating losses and negative cash flows from operations since inception. To date, the Corporation has financed its operations through the public offering and private placement of Common Shares, (“Common Shares”; with or without warrants) and convertible debt, the proceeds from research grants and research tax credits, and the exercises of warrants, rights and options. To achieve the objectives of its business plan, Acasti plans to raise the necessary funds through a possible combination of additional warrant exercises, debt facilities, securities offerings and the establishment of strategic alliances, as well as additional research grants and research tax credits. The ability of the Corporation to complete needed financing and ultimately achieve profitable operations is dependent on a number of factors outside of the Corporation’s control. See “Risk Factors” in this MD&A and in Acasti’s Annual Report on Form 20-F for the fiscal year ended March 31, 2019.
The Corporation has incurred operating losses and negative cash flows from operations since inception. The Corporation’s current assets of $27.8 million as at September 30, 2019 include cash and cash equivalents and marketable securities totaling $25.8 million. The Corporation’s current liabilities total $15.4 million at September 30, 2019 and are comprised primarily of amounts due to or accrued for creditors. Management projects that additional funds will be needed in the future, for activities necessary to prepare for commercial launch, including the scale up of our manufacturing operations, the completion of the potential regulatory (NDA) submission package (assuming positive Phase 3 clinical results), and the expansion of business development and US commercial launch activities. The Corporation is working towards development of strategic partner relationships, as well as actively seeking additional non-dilutive funds in the near future, but there can be no assurance as to when or whether Acasti will complete any strategic collaborations or non-dilutive financings. Consequently, the Corporation may need to raise additional equity capital in the future to fund these activities. In particular, raising additional capital is subject to market conditions not within the Corporation’s control. If the Corporation does not raise additional funds or find one or more strategic partners, it may not be able to realize its assets and discharge its liabilities in the normal course of business. As a result, there exists a material uncertainty that casts substantial doubt about the Corporation’s ability to continue as a going concern and, therefore, realize its assets and discharge its liabilities in the normal course of business.
The financial statements have been prepared on a going concern basis, which assumes the Corporation will continue its operations in the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the ordinary course of business. These financial statements do not include any adjustments to the carrying values and classification of assets and liabilities and reported expenses that may be necessary if the going concern basis was not appropriate for these financial statements. If the Corporation was unable to continue as a going concern, material write-downs to the carrying values of the Corporation’s assets, including the intangible asset, could be required.
20
SELECTED FINANCIAL INFORMATION
| Three-month periods ended | Six-month periods ended | |||||||||||||||
|
September 30, 2019 |
September 30, 2018 |
September 30, 2019 |
September 30, 2018 |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (28,343 | ) | (22,729 | ) | (40,144 | ) | (30,150 | ) | ||||||||
| Basic and diluted loss per share | (0.34 | ) | (0.62 | ) | (0.50 | ) | (0.88 | ) | ||||||||
| Non-IFRS operating loss1 | (6,672 | ) | (9,423 | ) | (16,332 | ) | (18,411 | ) | ||||||||
| Total assets | 37,840 | 19,920 | 37,795 | 19,920 | ||||||||||||
| Working capital2 | 12,395 | (5,813 | ) | 12,350 | (5,813 | ) | ||||||||||
| Total non-current financial liabilities | 24,137 | 21,570 | 24,137 | 21,570 | ||||||||||||
| Total equity | (1,670 | ) | (14,824 | ) | (1,715 | ) | (14,824 | ) | ||||||||
RECONCILIATION OF NET LOSS TO NON-IFRS OPERATING LOSS
| Three-month periods ended | Six-month periods ended | |||||||||||||||
|
September 30, 2019 |
September 30, 2018 |
September 30, 2019 |
September 30, 2018 |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (28,343 | ) | (22,729 | ) | (40,144 | ) | (30,150 | ) | ||||||||
| Add (deduct): | ||||||||||||||||
| Stock-based compensation | 1,298 | 326 | 1,623 | 577 | ||||||||||||
| Depreciation and amortization | 675 | 689 | 1,374 | 1,373 | ||||||||||||
| Financial expenses | 19,698 | 12,291 | 20,815 | 9,789 | ||||||||||||
| Non-IFRS operating loss | (6,672 | ) | (9,423 | ) | (16,332 | ) | (18,411 | ) | ||||||||
COMMENTS ON THE SIGNIFICANT VARIATIONS OF RESULTS FROM OPERATIONS FOR THE THREE AND SIX-MONTH PERIODS ENDED SEPTEMBER 30, 2019 AND 2018
The net loss of $28,343 or ($0.34) per share for the three months ended September 30, 2019 increased by $5,614 from the net loss for the three months ended September 30, 2018, while the loss per share decreased by ($0.28) per share from the loss of ($0.62) per share loss for the three months ended September 30, 2018. The per share loss decreased due to the issuance of 60,472,880 shares in relation mainly to the public financings that occurred in May and October of 2018 and the exercise of warrants during July and August 2019.
The increased net loss resulted primarily from a net financial expenses of $19,698 for the three months ended September 30, 2019, as compared to net financial expenses of $12,291 for the three months ended September 30, 2018, due mostly to the change in fair value of the warrant derivative liability partially offset by a decrease in the number of warrants. In addition, the increase in net loss is the result of creating our senior commercial team during the second quarter of fiscal year 2019 to support expanded business and development activities, and of additional administrative fees incurred in connection with the implementation of a new Enterprise Resource Planning (“ERP”) system and increased insurance cost, as well as increased accounting and legal fees. Furthermore, stock-based compensation expense increased by $972 as result of new grants. The increase in administrative and sales and marketing expenses were partially offset by a decrease on research and development expenses as the Phase 3 clinical trial program is getting closer to completion.
_______________________________
1 The Non-IFRS operating loss is not a standard measure endorsed by IFRS requirements. A reconciliation to the Corporation’s net loss is presented above.
2 The working capital is calculated by subtracting current liabilities from current assets. Because there is no standard method endorsed by IFRS requirements, the results may not be comparable to similar measurements presented by other public companies.
21
The net loss of $40,099 or ($0.50) per share for the six months ended September 30, 2019 increased by $9,949 from the net loss for the six months ended September 30, 2018, while the loss per share decreased by ($0.38) per share from the loss of ($0.88) per share loss for the six months ended September 30, 2018. The per share loss decreased due to the issuance of 60,472,880 shares in relation mainly to the public financings that occurred in May and October of 2018 and the exercise of warrants during July and August 2019.
The increased net loss resulted primarily from a net financial expenses of $20,815 for the six months ended September 30, 2019, as compared to net financial expenses of $9,789 for the six months ended September 30, 2018, due mostly to the change in fair value of the warrant derivative liability, partially offset by a decrease in the number of warrants. In addition, the increase in net loss is the result of creating our senior commercial team during the second quarter of fiscal year 2019 to support expanded business and development activities, and of additional administrative fees incurred in connection with the implementation of a new ERP system and increased insurance cost, as well as increased accounting and legal fees. Furthermore, stock-based compensation expense increased by $1,046 as result of new grants. The increase in administrative and sales and marketing expenses were partially offset by a decrease on research and development expenses as the Phase 3 clinical trial program is getting closer to completion.
22
Breakdown of major components of the statement of earnings and comprehensive loss.
| Research and development expenses | ||||||||||||||||
| Three Months Ended | Six Months Ended | |||||||||||||||
|
September 30, 2019 |
September 30, 2018 |
September 30, 2019 |
September 30, 2018 |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Salaries and benefits | 558 | 334 | 1,109 | 757 | ||||||||||||
| Research contracts | 3,289 | 7,926 | 9,951 | 15,143 | ||||||||||||
| Professional fees | 318 | 183 | 538 | 588 | ||||||||||||
| Other | 137 | 76 | 219 | 225 | ||||||||||||
| Government grants & tax credits | - | (110 | ) | (100 | ) | (180 | ) | |||||||||
| Sub-total | 4,302 | 8,409 | 11,717 | 16,533 | ||||||||||||
| Stock-based compensation | 271 | 30 | 374 | 109 | ||||||||||||
| Depreciation and amortization | 674 | 689 | 1,374 | 1,373 | ||||||||||||
| Total | 5,246 | 9,128 | 13,465 | 18,015 | ||||||||||||
| General and administrative expenses | ||||||||||||||||
| Three Months Ended | Six Months Ended | |||||||||||||||
|
September 30, 2019 |
September 30, 2018 |
September 30, 2019 |
September 30, 2018 |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Salaries and benefits | 480 | 373 | 954 | 790 | ||||||||||||
| Professional fees | 640 | 381 | 1,138 | 709 | ||||||||||||
| Other | 374 | 135 | 698 | 254 | ||||||||||||
| Sub-total | 1,494 | 889 | 2,790 | 1,753 | ||||||||||||
| Stock-based compensation | 883 | 257 | 1,073 | 429 | ||||||||||||
| Total | 2,377 | 1,146 | 3,863 | 2,182 | ||||||||||||
| Sales and Marketing Expenses | ||||||||||||||||
| Three Months Ended | Six Months Ended | |||||||||||||||
| September 30, 2019 |
September 30, 2018 |
September 30, 2019 |
September 30, 2018 |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Salaries and benefits | 347 | 121 | 600 | 121 | ||||||||||||
| Marketing | 246 | - | 737 | - | ||||||||||||
| Professional fees | 51 | - | 76 | - | ||||||||||||
| Other | 234 | 4 | 367 | 4 | ||||||||||||
| Sub-total | 878 | 125 | 1,780 | 125 | ||||||||||||
| Stock-based compensation | 144 | 39 | 176 | 39 | ||||||||||||
| Total | 1,022 | 164 | 1,956 | 164 | ||||||||||||
23
Three months ended September 30, 2019 compared to the three months ended September 30, 2018:
During the three months ended September 30, 2019, Acasti continued its planned advancement of the two-study TRILOGY Phase 3 clinical study program for its drug candidate, CaPre, in partnership with one of the world’s largest providers of biopharmaceutical development and clinical outsourcing services (“CRO”). R&D expenses before depreciation, amortization and stock-based compensation expense for the three months ended September 30, 2019 totaled $4,302, compared to $8,409 for the three months ended September 30, 2018. This $4,107 net decrease was mainly attributable to a $4,637 decrease in research contracts, partially offset by an increase in professional fees of $135 and an increase in salaries and benefits of $224 due to increased headcount. The lower research contract expense is attributed primarily to the advancement of the CRO as the Phase 3 clinical trial program is getting closer to completion.
G&A expenses totaling $1,494 before stock-based compensation expense for the three months ended September 30, 2019, and increased by $605 from $889 for the three months ended September 30, 2018. This increase was mainly attributable to a $178 increase associated with our Directors and Officers insurance policy, as well as an increase of $259 in corporate, accounting and legal fees. Salaries and benefits also increased due to higher headcount and higher bonus accrual.
S&M expenses were $878 before stock-based compensation expense for the three months ended September 30, 2019 compared to $125 for the three months ended September 30, 2018. During the second quarter ended September 30, 2018, we hired our Chief Commercial Officer to support expanded business, market development and product launch activities that required additional headcount, marketing expenses and consulting services.
The stock-based compensation expense increased by $972 to $1,298 for the three months ended September 30, 2019 from $326 for the three months ended September 30, 2018. The increase is mainly due to 2.1 million stock options granted to existing and new employees and directors during the six months ended September 30, 2019, partially offset by stock options exercised and forfeited. The weighted average fair value of the options granted to employees and directors during the six months ended September 30, 2019 was $1.88, and no options were granted to consultants.
The depreciation and amortization expense decreased by $16 to $675 for the three months ended September 30, 2019 from $689 for the three months ended September 30, 2018, remaining relatively constant.
Net financial expenses for the three months ended September 30, 2019 was $19,698 compared to a financial loss of $12,291 for the three months ended September 30, 2018. The net increase in loss of $7,407 was mainly attributable to an increase in loss from the changes in fair value of derivative warrant liabilities of $7,351, partially offset by a lower number of warrants as 5.8 million derivative warrant liabilities were exercised during July and August 2019.
Six months ended September 30, 2019 compared to the six months ended September 30, 2018:
During the six months ended September 30, 2019, Acasti continued its planned advancement of the two-study TRILOGY Phase 3 clinical study program for its drug candidate, CaPre, in partnership with one of the world’s largest providers of biopharmaceutical development and clinical outsourcing services (“CRO”). R&D expenses before depreciation, amortization and stock-based compensation expense for the six months ended September 30, 2019 totaled $11,717, compared to $16,533 for the six months ended September 30, 2018. This $4,816 net decrease was mainly attributable to a $5,197 decrease in research contracts, partially offset by an increase in salaries and benefits of $352 due to increased headcount and higher bonus accrual. The lower research contract expense is attributed primarily to the advancement of the CRO as the Phase 3 clinical trial program is getting closer to completion.
G&A expenses totaling $2,790 before stock-based compensation expense for the six months ended September 30, 2019, and increased by $1,037 from $1,753 for the six months ended September 30, 2018. This increase was mainly attributable to a $362 increase associated with our Directors and Officers insurance policy, as well as an increase of $429 in accounting, corporate and legal fees. Salaries and benefits also increased due to higher headcount and higher bonus accrual.
S&M expenses were $1,780 before stock-based compensation expense for the six months ended September 30, 2019 compared to $125 for the six months ended September 30, 2018. During the second quarter ended September 30, 2018, we hired our Chief Commercial Officer to support expanded business, market development and product launch activities that required additional headcount and marketing expenses, notably branding and consulting services.
24
The stock-based compensation expense increased by $1,046 to $1,623 for the six months ended September 30, 2019 from $577 for the six months ended September 30, 2018. The increase is mainly due to 2.1 million stock options granted to existing and new employees and directors, partially offset by stock options exercised and forfeited. The weighted average fair value of the options granted to employees and directors during the six months ended September 30, 2019 was $1.55, and no options were granted to consultants.
The depreciation and amortization expense remained constant.
Net financial expenses for the six months ended September 30, 2019 was $20,815 compared to net financial expenses of $9,789 for the six months ended September 30, 2018. The net increase in loss of $11,026 was mainly attributable to an increase in loss from the changes in fair value of derivative warrant liabilities of $11,829, partially offset by a decrease in financing fees of $653 incurred in connection with the financing completed in May 2018 and the exercise of 5.8 million derivative warrant liabilities during July and August 2019.
Two separate derivative warrant liabilities are included in the statement of financial position as at September 30, 2019, and September 30, 2018. These derivative warrant liabilities stem from the financing transactions that took place in May 2018 and December 2017. The derivative warrant liabilities are re-measured at each reporting date using the Black-Scholes option pricing model. The valuations are driven by the fluctuation in the Corporation’s stock price resulting in an increased or decreased loss or gain related to the change in fair value of the warrant liabilities and increasing or decreasing the corresponding liability in the statement of financial position.
25
SELECTED QUARTERLY FINANCIAL DATA
| September 30, | June 30, | March 31, | December 31, | |||||||||||||
| 2019 | 2019 | 2019 | 2018 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (28,343 | ) | (11,756 | ) | (16,806 | ) | (4,610 | ) | ||||||||
| Add (deduct): | ||||||||||||||||
| Depreciation and amortization | 675 | 699 | 731 | 723 | ||||||||||||
| Stock based compensation | 1,298 | 325 | 128 | 336 | ||||||||||||
| Shares issued as legal settlement | - | - | 990 | - | ||||||||||||
| Financial expense (income) | 19,698 | 1,117 | 2,862 | (6,100 | ) | |||||||||||
| Non-IFRS operating loss | (6,672 | ) | (9,615 | ) | (12,095 | ) | (9,651 | ) | ||||||||
| Basic and diluted net loss per share | (0.08 | ) | (0.12 | ) | (0.22 | ) | (0.07 | ) | ||||||||
| September 30, | June 30, | March 31, | December 31, | |||||||||||||
| 2018 | 2018 | 2018 | 2017 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (22,729 | ) | (7,421 | ) | (8,140 | ) | (6,079 | ) | ||||||||
| Add (deduct): | ||||||||||||||||
| Depreciation and amortization | 689 | 684 | 667 | 671 | ||||||||||||
| Stock based compensation | 326 | 251 | 268 | 330 | ||||||||||||
| Financial expense (income) | 12,291 | (2,502 | ) | 778 | 929 | |||||||||||
| Non-IFRS operating loss | (9,423 | ) | (8,988 | ) | (6,427 | ) | (4,149 | ) | ||||||||
| Basic and diluted net loss per share | (0.62 | ) | (0.23 | ) | (0.32 | ) | (0.40 | ) | ||||||||
The quarterly year-to-year non-IFRS operating loss variances are mainly attributable to fluctuations in R&D expenses associated with its TRILOGY Phase 3 clinical trial program from quarter-to-quarter, as well as an increase in G&A and S&M expenses over the last four quarters as the Corporation increased back-office administrative resources and added new commercial team members to manage the commercialization and marketing efforts.
26
LIQUIDITY AND CAPITAL RESOURCES
Share Capital Structure
The Corporation’s authorized share capital consists of an unlimited number of Class A, Class B, Class C, Class D and Class E shares, without par value. Issued and outstanding fully paid shares, stock options, restricted shares units and warrants, were as follows for the periods ended:
| September 30, 2019 |
March 31, 2019 |
|||||||
|
Number outstanding |
Number outstanding |
|||||||
| Class A shares, voting, participating and without par value | 85,188,095 | 78,132,734 | ||||||
| Stock options granted and outstanding | 6,178,128 | 4,046,677 | ||||||
| May 2018 public offering of warrants exercisable at $1.31, until May 9, 2023 | 6,668,750 | 10,188,100 | ||||||
| Public offering broker warrants May 2018 exercisable at $1.05 until May 9, 2023 | 422,976 | 547,975 | ||||||
| December 2017 U.S. public offering of warrants exercisable at US$1.26, until December 27, 2022 |
7,476,770 | 9,801,861 | ||||||
| December 2017 U.S. broker warrants exercisable at US$1.2625, until December 27, 2022 | 259,121 | 495,050 | ||||||
| February 2017 public offering of warrants exercisable at $2.15, | ||||||||
| until February 21, 2022 | 1,818,034 | 1,904,034 | ||||||
| 2017 unsecured convertible debentures conversion option | ||||||||
| contingent warrants exercisable at $1.90, until February 21, 20203 | 1,052,630 | 1,052,630 | ||||||
| Total fully diluted shares | 109,064,504 | 106,169,061 | ||||||
Cash Flows and Financial Condition between the three and six-month periods ended September 30, 2019 and 2018
Summary
As at September 30, 2019, cash and cash equivalents totaled $25,812 with a net increase of cash totaling $4,891 and $3,291 for the three and six-month periods ended September 30, 2019, respectively. This compares to cash and cash equivalents totaling $5,969 at September 30, 2018, and a use of cash totaling $6,891 and $2,254 for the three and six-month periods ended September 30, 2018, respectively.
Operating activities
During the three months ended September 30, 2019 and September 30, 2018, the Corporation’s operating activities used cash of $8,276 and $6,550, respectively, and during the six-months periods ended September 30, 2019 and September 30, 2018, the Corporation’s operating activities used cash of $17,480 and $12,180, respectively further modified by changes in working capital, excluding cash. The difference between the use of cash flows in operating activities for the three and six-months periods ended September 30, 2019 and 2018, when compared to the net losses for each period is mainly attributable to the change in non-cash expenses, (see “Reconciliation of Net Loss to Non-IFRS Operating Loss”), further modified by changes in working capital, excluding cash.
____________________________
3 The debentures are convertible into Common Shares at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures. Should the Corporation pay all or any portion of the convertible debentures before maturity, then warrants become exercisable at $1.90 per Common Share for the equivalent convertible debenture amount prepaid.
27
Investing activities
During the three months ended September 30, 2019, the Corporation’s investing activities provided cash of $4,578, primarily due to the maturity of marketable securities, compared to a use of cash of $194 for the three months ended September 30, 2018, due to the acquisition of equipment of $257, offset by interest received of $63.
During the six months ended September 30, 2019, the Corporation’s investing activities provided cash of $12,113 primarily due to the maturity of marketable securities, compared to a use of cash of $464 for the six months ended September 30, 2018 due to the acquisition of equipment of $544, offset by interest received of $80.
Financing activities
During the three months ended September 30, 2019, the Corporation’s financing activities provided cash totaling $8,673 due primarily to the exercise of warrants. During the three months ended September 30, 2018, the Corporation used cash of $126 for payment of transaction costs related to the public offering.
During the six months ended September 30, 2019, the Corporation’s financing activities generated cash of $8,661 mainly from the exercise of warrants, and during the six months ended September 30, 2018, the Corporation’s financing activities generated cash of $10,311 mainly from the net proceeds of the public offering.
ATM Program
On February 14, 2019, the Corporation entered into an “at-the-market” (“ATM”) sales agreement with B. Riley FBR, Inc., pursuant to which the Corporation’s common shares may be sold from time to time for aggregate gross proceeds of up to US $30 million, with sales only being made on the NASDAQ Stock Market. The common shares may be distributed at market prices prevailing at the time of any sale and, as a result, prices may vary between purchasers and during the period of distribution. As at September 30, 2019, no securities have been issued in relation to the ATM. Costs incurred in connection to the ATM of $224 have been recorded as deferred financing costs.
October 2018 Public Offering
On October 9, 2018, the Corporation closed a U.S. public offering of 16,600,000 Common Shares at a price of US$1.00 per share. In addition, the underwriters fully exercised their over-allotment option to purchase 2,490,000 additional Common Shares at the same public offering price. This offering generated gross proceeds of $24.7 million (US$19.1 million), which resulted in net proceeds to the Corporation of $22.6 million (US$17.4 million) and a total of 19,090,000 Common Shares issued.
On October 23, 2018, the Corporation closed a Canadian public offering of 18,750,000 Common Shares at a price of $1.28 per share. In addition, the underwriters fully exercised their over-allotment option to purchase 2,812,500 additional Common Shares at the same public offering price. This offering generated gross proceeds of $27.6 million, which resulted in net proceeds to the Corporation of approximately $25.4 million and a total of 21,562,500 Common Shares issued.
May 2018 Public Offering
On May 9, 2018 the Corporation closed a Canadian public offering issuing 9,530,000 units of Acasti (“Units”) at a price of $1.05 per Unit for gross proceeds of $10 million. The units issued consist of 9,530,000 Common Shares and 9,530,000 Warrants. Each Warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of $1.31 at any time until May 9, 2023.
On May 14, 2018, the underwriters exercised their over-allotment option by purchasing an additional 1,429,500 units at a price of $1.05 per Unit, for additional gross proceeds of $1.5 million. The units issued consist of 1,429,500 Common Shares and 1,429,500 warrants. Each Warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of $1.31 at any time until May 9, 2023.
28
The warrant component of these Units are Derivative Warrant Liabilities for accounting purposes due to the warrant agreement, which contains certain contingent provisions that allow for cash settlement. The proceeds of the offering are required to be split between the Derivative Warrant Liabilities and the equity-classified Common shares at the time of issuance of the Units. The fair value of the Derivative Warrant Liabilities at the time of issuance was determined to be $4.3 million and the residual of the proceeds of $6.2 million was allocated to the Common Shares. Issuance costs related to this transaction totaled approximately $1.8 million and have been allocated between the Derivative Warrant Liabilities and Common shares based on relative value. Resulting from this allocation, $0.7 million has been allocated to the Derivative Warrant Liability and is recognized in finance costs in the Statements of Earnings and Comprehensive Loss, whereas the remaining portion of $1.1 million in issuance costs was allocated to the Common Shares and recognized as a reduction to share capital, in the Statements of Financial Position.
The weighted average fair value of the 2018 Warrants issued in May 2018 was determined to be $0.39 per warrant. Changes in the fair value of the 2018 Warrants are recognized in finance expense.
As part of the May financing, the Corporation also issued broker warrants to purchase up to 547,975 Common Shares. Each broker warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of $1.05, at any time until May 9, 2023. The broker warrants are considered for compensation to non-employees under IFRS 2, stock-based compensation, and are accounted for at fair value at issuance date and not subsequently revalued.
29
Financial Position
The following table details the significant changes to the statements of financial position as at September 30, 2019 compared to the prior fiscal period end at March 31, 2019:
| Accounts | Increase (Decrease) |
Comments | ||||
| Cash and cash equivalents | 3,291 | See cash flow statement | ||||
| Marketable securities | (11,866 | ) | Progression of research contracts | |||
| Receivable | (314 | ) | Timing of receipts | |||
| Deferred Financing Costs | 45 | Additional accounting and legal fees incurred in connection with the ATM program | ||||
| Prepaid expenses | (746 | ) | Progression of research contracts | |||
| Equipment | 121 | Depreciation net of acquisition of equipment | ||||
| Intangible asset | (1,162 | ) | Amortization | |||
| Trade and other payables | (2,975 | ) | Timing of payments net of accruals and settlement of provision for legal settlement via the issuance of common shares | |||
| Derivative warrant liabilities | 7,874 | Change in fair value and exercise of derivative warrants | ||||
| Unsecured convertible debentures | 102 | Accretion of interest | ||||
See the statement of changes in equity in the Corporation’s financial statements for details of changes to the equity accounts since September 30, 2019.
Treasury Operations
The Corporation’s treasury policy is to invest cash that is not required immediately into instruments with an investment strategy based on capital preservation. Cash equivalents and marketable securities are primarily made in guaranteed investment certificates (GICs), term deposits and high-interest savings accounts, which are issued and held with Canadian chartered banks; high rated promissory notes issued by government bodies and commercial paper. The Corporation holds cash denominated in both US and CAD dollars. Funds received in US dollars from the equity private placement are invested as per the Corporation’s treasury policy in US dollar investments and converted to CAD dollars as appropriate to fulfill operational requirements and funding.
Derivative warrant liabilities
The 10,188,100 Warrants issued as part of the Corporation’s May 2018 public offering were recognized as Derivative Warrant Liabilities with a fair value of $4,272. During the six months ended September 30, 2019, a total of 3,519,350 warrants were exercised. As of September 30, 2019, the Derivative Warrant Liability for the remaining 6,668,750 Warrants totaled $11,384, which represents the fair value of these warrants. The weighted average fair value of the 2018 Warrants issued was determined to be $0.39 per warrant at inception and approximately $1.71 per warrant as at September 30, 2019.
On December 27, 2017, 9,801,861 warrants were issued as part of the Corporation’s U.S. public offering and recognized as Derivative Warrant Liabilities with a fair value of $5,873 (“2017 Warrants”). The 2017 Warrants are Derivative Warrant Liabilities for accounting purposes due to the currency of the exercise price (US$) being different from the Corporation’s Canadian dollar functional currency. During the six months ended September 30, 2019, 2,325,091 warrants were exercised (including 288,217 warrants exercised in a cashless manner). The fair value of the 2017 Warrants as of September 30, 2019, totaled $12,753. The fair value of the 2017 Warrants was determined to be $0.60 per warrant upon issuance and approximately $1.71 per warrant as of September 30, 2019.
The increase in the fair value of both existing derivative warrant liabilities as at September 30, 2019 is due to the increase in the Corporation’s share price and the dilution factor.
30
During October 2018, 771,400 - 2018 Warrants were exercised with one Common Share of the Corporation acquired at an exercise price of $1.31 and aggregate gross proceeds of approximately $1.0 million. In addition, 4,455, of the 2017 Warrants were exercised in a cashless manner to acquire 1,074 Common Shares of the Corporation. A total of 772,474 Common Shares were issued as a result of 775,855 warrants exercised.
During the period from June to September 2019, the following warrants were exercised with the resulting cash proceeds:
|
Number exercised |
Proceeds | |||||||
| $ | ||||||||
| May 2018 over-allotment Warrants 2018 | 3,519,350 | 4,610 | ||||||
| Series December 2017 US Public offering Warrants 2017 | 2,272,803 | 3,798 | ||||||
| Public offering warrants February 2017 | 86,000 | 185 | ||||||
| Public offering Broker warrants May 2018 | 125,000 | 131 | ||||||
| 6,003,153 | 8,724 | |||||||
In addition, 235,929, broker warrants and 52,288 derivative warrants offered as part of the December 2017 U.S. public offering were exercised in a cashless manner to acquire 136,013 Common Shares of the Company.
Contractual Obligations, Off-Balance-Sheet Arrangements and Commitments
As at September 30, 2019, the Corporation’s liabilities total $39,510, of which $15,373 is due within twelve months, $24,137 relates to Derivative Warrant Liabilities that will be settled in Common Shares and $1,919 of outstanding unsecured convertible debentures is also projected to be settled in Common Shares. However, the principal amount of unsecured convertible debentures may be prepaid, in whole or in part, at any time and from time to time, in cash, at the sole discretion of the Corporation. The debentures are convertible into Common Shares at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures.
A summary of the contractual obligations at September 30, 2019, is as follows:
| Carrying value | Total contractual cash flows | 1 year or less | 1 to 3 years | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Trade, other payables and due to related party | 13,454 | 13,454 | 13,454 | - | ||||||||||||
| Lease | 39 | 39 | 39 | - | ||||||||||||
| Unsecured convertible debentures | 1,919 | 2,000 | 2,000 | - | ||||||||||||
| Total | 15,412 | 15,493 | 15,493 | - | ||||||||||||
The Corporation has no off-balance sheet arrangements.
Research and development contracts and contract research organizations agreements
The Corporation utilizes Contract Manufacturing Organizations (CMOs) for the development and production of clinical materials and contract research organizations to perform services related to the Corporation’s clinical trials. Pursuant to the agreements with these CMOs and contract research organizations, the Corporation has either the right to terminate the agreements without penalties or under certain penalty conditions.
31
Lease
During Fiscal 2018, the Corporation entered into a lease agreement, for its research and development and quality control laboratory facility located in Sherbrooke, Québec, resulting in a commitment of $39 over the remaining six months of the lease term.
Contingencies
The Corporation evaluates contingencies on an ongoing basis and establishes loss provisions for matters in which losses are probable and the amount of the loss can be reasonably estimated.
On May 10, 2019, the Corporation announced the settlement regarding legal claims made by its former chief executive (“CEO”) officer with respect to the termination of his employment. Pursuant to the settlement agreement, the Corporation agreed to issue 900,000 common shares at $1.10 per share to the former CEO. In addition, the Corporation agreed to reimburse the former CEO for legal fees of $64. Furthermore, pursuant to the settlement agreement, the Corporation received a full and final release from the former CEO on all procedures in connection with the termination of his employment. This settlement was accrued as a short-term liability as at March 31, 2019 and the expense of $1,054 was included as part of General and administrative expenses. During May 2019, the shares were issued and the liability of $990 reclassified as Equity.
Related Party Transactions
Neptune Wellness Solutions (formerly Neptune Technologies –“Neptune”) Acasti’s former parent company, is no longer a related party of the Corporation. Neptune’s ownership was reduced below a control and significant influence position, following Acasti’s U.S. public financing activities in December 2017 and January 2018.
Acasti was charged by Neptune, the Corporation’s former parent Company, for certain costs incurred by Neptune for the benefit of the Corporation. Over the last year, the Corporation has almost fully eliminated its dependence on the support of Neptune for its G&A needs and in consequence, the Corporation is now incurring incremental costs because of increased administrative headcount.
During the three and six-months period ended September 30, 2019, the Corporation recognized expenses of $45 and $86, respectively, in G&A expenses in relation to supplies and incremental costs, compared to $57 and $130, for three and six-months period ended September 30, 2018, respectively. The account payable to Neptune amounted to $17 at September 30, 2019, and $2 at March 31, 2019.
The key management personnel are the officers of the Corporation and the members of the Board of Directors of the Corporation. They control collectively less than 1% of the voting shares of the Corporation. See note 6 to the financial statements for disclosures of key management personnel compensation.
Controls and procedures
In accordance with the Canadian Securities Administrators’ National Instrument 52-109, the Corporation has filed certificates signed by the Corporation’s Chief Executive Officer (“CEO”) and Vice President Finance that among other things, report on the design of disclosure controls and procedures and the design of internal control over financial reporting.
Disclosure controls and procedures
The CEO and Vice President Finance, have designed disclosure controls and procedures, or has caused them to be designed under their supervision, in order to provide reasonable assurance that:
| · | material information relating to the Corporation has been made known to them; and |
| · | information required to be disclosed in the Corporation’s filings is recorded, processed, summarized and reported within the time periods specified in securities legislation. |
32
Changes in internal control over financial reporting (“ICFR”)
During the six months ended September 30, 2019, we implemented a new fully integrated, scalable, and user-friendly Enterprise Resource Planning (“ERP”) system “Navision”, to replace our former accounting system. Navision went live on April 1, 2019 and it has been used to record our transactions and produce our financial and operating reports. Certain of the Corporation’s controls have been amended to reflect the new ERP system.
There have been no other changes in the Corporation’s ICFR during the three months ended September 30, 2019 that have materially affected, or are reasonably likely to materially affect its ICFR.
Upcoming Changes in IFRS / Foreign Private Issuer Status
Under the Securities Exchange Act of 1934 (“Exchange Act”), a foreign private issuer (“FPI”) is an entity incorporated or organized under the laws of a jurisdiction outside of the U.S., unless:
| • | more than 50% of its outstanding voting securities are directly or indirectly owned of record by U.S. residents; and |
| • | any of the following applies: (i) the majority of its executive officers or directors are U.S. citizens or residents; (ii) more than 50% of its assets are located in the U.S.; or (iii) its business is administered principally in the U.S. |
A corporation’s ongoing FPI status is tested annually at the end of the most recently completed second fiscal quarter. If an issuer fails to qualify as an FPI at the end of its second fiscal quarter, it remains eligible to use the forms and rules applicable to FPIs until the end of that financial year. Historically, we met the definition of an FPI, and as such, prepared consolidated financial statements in accordance with IFRS, reported with the United States Securities and Exchange Commission (“SEC”) on FPI forms, and complied with SEC rules and regulations applicable to FPIs.
As of September 30, 2019, due to an increase in the percentage of its Common Shares held by U.S. residents
above 50%, the Corporation has determined that it no longer qualifies as an FPI. Therefore, the Corporation plans to transition
to U.S. domestic corporation reporting status and become subject to SEC reporting requirements applicable to a U.S. domestic corporation,
beginning on April 1, 2020. These reporting requirements will require that the Corporation’s fiscal 2020 financial statements
be recast into United States Generally Accepted Accounting Principles for all periods presented, which will include the fiscal
2019 comparative financial information. Therefore, the last period under which the Corporation will report under IFRS is the third
quarter ended December 31, 2019. The extent of the impact of this transition has not yet been determined. In addition, the Corporation
will be required to file Form 10-K annual, Form 10-Q quarterly and Form 8-K current reports with the SEC, its insiders will have
to comply with U.S. insider filing requirements under the Exchange Act, and the Corporation will
become subject to the SEC’s proxy rules.
33
RISK FACTORS
Investing in Acasti’s securities involves a high degree of risk due to, among other things, the nature of our business and the present stage of our development. Prospective and current investors should carefully consider the following risks and uncertainties, together with all other information in this MD&A, as well as our financial statements and the risks described in more detail in Item 3. “Risk Factors” and “Item 5. Operating and Financial Review and Prospects” in our Annual Report on Form 20-F for the fiscal year ended March 31, 2019 and our other public filings. If any of these risks actually occur, Acasti’s business, financial condition, prospects, results of operations or cash flow could be materially and adversely affected and all or a part of the value of an investor’s investment in Acasti can be lost. Additional risks or uncertainties not currently known to Acasti, or that we currently deem immaterial, may also negatively affect our business operations.
General Risks Related to the Corporation
| · | We may not be able to maintain our operations and advance our research and development of CaPre without additional funding. |
| · | We may never become profitable or be able to sustain profitability. |
| · | If outcome studies being conducted by our competitors testing the impact of OM3 on treating patients with high TGs are negative, there could also be an adverse impact for CaPre. |
| · | We are and will continue to rely on third parties to conduct our TRILOGY Phase 3 program for CaPre. |
| · | We rely on third parties to manufacture, produce and supply CaPre and we may be adversely affected if those third parties are unable or unwilling to fulfill their obligations, including complying with FDA requirements. |
| · | We have historically had no marketing and sales organization and, as a corporation, no history in marketing products. If we are unable to properly establish marketing and sales capabilities or enter into agreements with a strategic partner to market and sell CaPre, we may not be able to generate revenue. |
| · | If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy. |
| · | Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses. |
| · | Our prospects currently depend entirely on the success of CaPre, which is still in clinical development, and we may not be able to generate revenues from CaPre. |
| · | We may not be able to obtain required regulatory approvals for CaPre. |
| · | Even if we receive regulatory approval for CaPre, it may just be for a limited indication. |
| · | We may be unable to find successful strategic partnerships to develop and commercialize CaPre. |
| · | We may be unable to develop alternative product candidates. |
| · | We may not be able to compete effectively against our competitors’ pharmaceutical products. |
| · | CaPre could face competition from products for which no prescription is required. |
| · | Recent and future legal developments could make it more difficult and costly for us to obtain regulatory approvals for CaPre and negatively affect the prices we may charge. |
| · | Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance. If there is not sufficient reimbursement for CaPre, it is less likely that it will be widely used. |
| · | Even if we obtain FDA approval of CaPre, we may never obtain approval or commercialize it outside of the United States, which would limit our ability to realize CaPre’s full market potential. |
| · | If we or our third-party service providers fail to comply with healthcare laws and regulations or government price reporting laws, we could be subject to civil or criminal penalties. |
34
| · | The research, development and manufacture of CaPre involves using potentially hazardous materials. |
| · | Interruptions of our supply of CaPre could disrupt our planned TRILOGY Phase 3 program and, if CaPre reaches commercialization, impair any future revenue streams. |
| · | If product liability lawsuits are brought against us, we may incur substantial liabilities and be required to cease the sale, marketing and distribution of CaPre. We may not achieve our publicly announced milestones on time, or at all. |
| · | We may be subject to foreign exchange rate fluctuations. |
| · | While there is no indication of this in previous studies, CaPre may cause or be perceived to cause undesirable side effects or have other properties that could delay or prevent its regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any. |
Risks Related to Intellectual Property
| · | In addition to our own patents, CaPre is covered by patents that are sublicensed to us by Neptune. |
| · | It is difficult and costly to protect our intellectual property rights. |
| · | CaPre may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development and commercialization efforts. |
| · | If we do not protect our trademark for CaPre, we may not be able to build name recognition in our markets of interest. |
| · | We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful. |
| · | Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect CaPre and any of our other future product candidates. |
| · | We may not be able to protect our intellectual property rights throughout the world. |
Risks Relating to Our Common Shares
| · | The price of our common shares may be volatile. |
| · | Forward-Looking Statements may prove to be inaccurate. |
| · | Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or product candidates. |
| · | There can be no assurance that an active market for our common shares will be sustained. |
| · | A large number of common shares may be issued and subsequently sold upon the exercise of existing warrants. The sale or availability for sale of existing warrants or other securities convertible in common shares may depress the price of our common shares. |
| · | We do not currently intend to pay any cash dividends on our common shares in the foreseeable future. |
| · | If we fail to meet applicable listing requirements, the NASDAQ Stock Market or the TSXV may delist our common shares from trading, in which case the liquidity and market price of our common shares could decline. |
| · | We may pursue opportunities or transactions that adversely affect our business and financial condition. |
| · | As a “non-accelerated filer”, we are not required to comply with the auditor attestation requirements of the Sarbanes-Oxley Act. |
| · | U.S. investors may be unable to enforce certain judgments. |
35
Additional Information
Updated and additional information about the Corporation is available on SEDAR at www.sedar.com or on EDGAR at www.sec.gov/edgar.shtml.
As at November 12, 2019, the Corporation’s capital structure is as follows:
| Number outstanding | |
| Class A shares, voting, participating and without par value | 85,325,694 |
| Stock options granted and outstanding | 6,178,128 |
| May 2018 public offering of warrants exercisable at $1.31, until May 9, 2023 | 6,668,750 |
| Public offering broker warrants May 2018 exercisable at $1.05 until May 9, 2023 | 422,976 |
| December 2017 U.S. public offering of warrants exercisable at US$1.26, until December 27, 2022 | 7,346,271 |
| December 2017 U.S. broker warrants exercisable at US$1.2625, until December 27, 2022 | 259,121 |
| February 2017 public offering of warrants exercisable at $2.15 until February 21, 2022 | 1,810,934 |
| 2017 unsecured convertible debentures conversion option | |
| contingent warrants exercisable at $1.90, until February 21, 20204 | 1,052,630 |
| Total fully diluted shares | 109,064,504 |
___________________________
4 The debentures are convertible into Common Shares at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures. Should the Corporation pay all or any portion of the convertible debenture before maturity, then warrants become exercisable at $1.90 per Common Share for the equivalent convertible debenture amount prepaid.
36