20-F: Annual and transition report of foreign private issuers pursuant to Section 13 or 15(d)
Published on June 29, 2018
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
| ☐ | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) or (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| OR | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the fiscal year ended March 31, 2018 | |
| OR | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| OR | |
| ☐ | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 001-35776
Acasti Pharma Inc.
(Exact name of Registrant as specified in its charter)
N/A
(Translation of Registrant’s name into English)
Québec, Canada
(Jurisdiction of incorporation or organization)
545, Promenade du Centropolis, Suite 100, Laval, Québec H7T 0A3
(Address of principal executive office)
Linda P. O’Keefe, Chief Financial Officer Acasti Pharma Inc.
545, Promenade du Centropolis, Suite 100 Laval, Québec H7T 0A3
Tel: 450-687-2262
Fax: 450-687-2272
(Name, Telephone, Email and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class | Name of each exchange on which registered | |
| Common Shares, no par value | The NASDAQ Capital Market |
Securities registered or to be registered pursuant to Section 12(g) of the Act.
Not applicable
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
25,638,215 Common Shares issued and outstanding as of March 31, 2018.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ☐ Accelerated filer ☐ Non-accelerated filer ☐ Emerging growth company ☒
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act ☐.
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP ☐ | International Financial Reporting Standards as issued by the International Accounting Standards Board ☒ |
Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. Item 17 ☐ Item 18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
TABLE OF CONTENTS
INTRODUCTION AND USE OF CERTAIN TERMS
As used in this annual report on Form 20-F, or this annual report, unless the context otherwise requires, references to “we”, “our”, “us”, “Acasti”, “Acasti Pharma”, “Corporation”, “it”, “its” or similar terms refer to Acasti Pharma Inc.
Market data and certain industry data and forecasts included in this annual report were obtained from internal company surveys, market research, publicly available information, reports of governmental agencies and industry publications and surveys. We have relied upon industry publications as our primary sources for third-party industry data and forecasts. Industry surveys, publications and forecasts generally state that the information they contain has been obtained from sources believed to be reliable, but that the accuracy and completeness of that information is not guaranteed. We have not independently verified any of the data from third-party sources or the underlying economic assumptions they made. Similarly, internal surveys, industry forecasts and market research, which we believe to be reliable based upon our management’s knowledge of our industry, have not been independently verified. Our estimates involve risks and uncertainties, including assumptions that may prove not to be accurate, and these estimates and certain industry data are subject to change based on various factors, including those discussed under “Risk Factors” in this annual report. While we believe our internal business research is reliable and the market definitions we use in this annual report are appropriate, neither our business research nor the definitions we use have been verified by any independent source. This annual report may only be used for the purpose for which it has been published.
We own or have rights to trademarks, service marks or trade names that we use in connection with the operation of our business. In addition, our name, logo and website names and addresses are our service marks or trademarks. CaPre® is our registered trademark. The other trademarks, trade names and service marks appearing in this annual report are the property of their respective owners. Solely for convenience, the trademarks, service marks, tradenames and copyrights referred to in this annual report are listed without the ©, ® and TM symbols, but we will assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensors to these trademarks, service marks and tradenames.
Financial Information
All financial information in this annual report is presented in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB, unless otherwise specified.
We use multiple financial measures for the review of our operating performance. These measures are generally IFRS financial measures, but one adjusted financial measure, Non-IFRS operating loss (adding to net loss, finance expenses, depreciation and amortization and impairment loss, change in fair value of derivative warrant liabilities, stock-based compensation and by subtracting finance income and deferred income tax recovery), is also used to assess our operating performance. This non-IFRS financial measure is derived from our financial statements and is presented in a consistent manner. We use this measure, in addition to the IFRS financial measures, for the purposes of evaluating our historical and prospective financial performance, as well as our performance relative to competitors. All of these measures also help us to plan and forecast future periods as well as to make operational and strategic decisions. We believe that providing this Non-IFRS information to investors, in addition to IFRS measures, allows them to see our results through the eyes of our management, and to better understand our historical and future financial performance. See “Item 5. Operating and Financial Review and Prospects”, including for a reconciliation to net loss.
In this annual report, all references to “CA$” or “$” are to Canadian dollars, unless expressly otherwise stated. All amounts related to our financial results are presented in thousands of Canadian dollars, except where noted and per share amounts.
| - 1 - |
Exchange Rate Information
The following table presents the average exchange rate for one Canadian dollar expressed as one U.S. dollar for each of our last five fiscal years. The average rate is calculated using the average of the exchange rates on the last day of each month during the period.
| Fiscal Year Ended | Average | |||
| (US$) | ||||
| February 28,2014 | 0.9555 | |||
| February 28, 2015 | 0.8003 | |||
| February 29, 2016 | 0.7645 | |||
| March 31, 2017 | 0.7618 | |||
| March 31, 2018 | 0.7752 | |||
The following table presents the high and low exchange rate for one Canadian dollar expressed as one U.S. dollar for each month during the previous six months.
| Month | Low | High | ||||||
| (US$) | ||||||||
| November 2017 | 0.7759 | 0.7885 | ||||||
| December 2017 | 0.7760 | 0.7971 | ||||||
| January 2018 | 0.7978 | 0.8135 | ||||||
| February 2018 | 0.7807 | 0.8138 | ||||||
| March 2018 | 0.7641 | 0.7794 | ||||||
| April 2018 | 0.7747 | 0.7967 | ||||||
| May 2018 | 0.7680 | 0.7828 | ||||||
The exchange rates are based upon the daily average closing rate, as quoted by the Bank of Canada. As of June 28, 2018, the exchange rate for one Canadian dollar expressed as one U.S. dollar, as quoted by the Bank of Canada was $1.00 = US$0.7537.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains information that may be forward-looking information within the meaning of Canadian securities laws and forward-looking statements within the meaning of U.S. federal securities laws, both of which we refer to in this annual report as forward-looking information. Forward-looking information can be identified by the use of terms such as “may”, “will”, “should”, “expect”, “plan”, “anticipate”, “believe”, “intend”, “estimate”, “predict”, “potential”, “continue” or other similar expressions concerning matters that are not statements about the present or historical facts. Forward-looking information in this annual report includes, among other things, information or statements about:
| · | our ability to conduct all required clinical and nonclinical trials for CaPre, including the timing and results of those trials; |
| · | our strategy, future operations, prospects and the plans of our management; |
| · | the design, regulatory plan, timeline, costs and results of our clinical and nonclinical trials for CaPre; |
| · | the timing and outcome of our meetings and discussions with the U.S. Food and Drug Administration, or FDA; |
| · | our planned regulatory filings for CaPre, and their timing; |
| · | our expectation that our Bridging Study (as defined below) results will support our plan to get authorization from the FDA to use the 505(b)(2) pathway with new chemical entity, or NCE, status towards a New Drug Application, or NDA, approval in the United States; |
| · | the timing and results from two competitor outcomes studies in patients with high TGs (blood levels between 200-499 mg/dL); |
| · | the potential benefits and risks of CaPre as compared to other products in the pharmaceutical, medical food and natural health products markets; |
| · | our estimates of the size of the potential market for CaPre, unmet medical needs in that market, the potential for market expansion, and the rate and degree of market acceptance of CaPre if it reaches commercialization, and our ability to serve that market; |
| · | our anticipated marketing advantages and product differentiation of CaPre and its potential to become a best-in-class OM3 compound for the treatment of HTG; |
| · | the potential to expand CaPre’s indication for the treatment of high TGs (200-500 mg/dL); |
| · | the degree to which physicians would switch their patients to a product with CaPre’s target product profile; |
| · | our strategy and ability to develop, commercialize and distribute CaPre in the United States and elsewhere; |
| · | the manufacturing scale-up of CaPre beyond 20 tons and the related timing; |
| - 2 - |
| · | our ability to strengthen our patent portfolio and other means of protecting our intellectual property rights, including our ability to obtain additional patent protection for CaPre; |
| · | our expectation that following expiration of the license agreement with Neptune we will not require any license from third parties to support the commercialization of CaPre; |
| · | the availability, consistency and sources of our raw materials, including krill oil; |
| · | our expectation to be able to rely on third parties to manufacture CaPre whose manufacturing processes and facilities are in compliance with current good manufacturing practices, or cGMP; |
| · | the potential for OM3s in other cardiovascular medicine, or CVM, indications; |
| · | our intention and ability to build a US commercial organization and to successfully launch CaPre and compete in the US market; |
| · | our intention and ability to complete development and/or distribution partnerships to support the commercialization of CaPre outside of the US, and to pursue strategic opportunities to provide capital and market access; |
| · | our ability to reach a definitive agreement based upon a non-binding term sheet with a leading China-based pharmaceutical company for the commercialization of CaPre in certain Asian jurisdictions; |
| · | our need for additional financing and our estimates regarding our future financing and capital requirements; |
| · | our expectation regarding our financial performance, including our revenues, profitability, research and development, costs and expenses, gross margins, liquidity, capital resources, and capital expenditures; and |
| · | our projected capital requirements to fund our anticipated expenses, including our research and development and general and administrative expenses, and capital expenditures. |
Although the forward-looking information in this annual report is based upon what we believe are reasonable assumptions, you should not place undue reliance on that forward-looking information since actual results may vary materially from it. Important assumptions by us when making forward-looking statements include, among other things, assumptions by us that:
| · | we successfully and timely complete all required clinical and nonclinical trials necessary for regulatory approval of CaPre; |
| · | we successfully enroll and randomize patients in our TRILOGY Phase 3 program; |
| · | the timeline and costs for our clinical and nonclinical programs are not materially underestimated or affected by unforeseen circumstances; |
| · | CaPre is safe and effective; |
| · | outcome study data from two of our competitors in high HTG patients is positive; |
| · | we obtain and maintain regulatory approval for CaPre on a timely basis; |
| · | we are able to attract, hire and retain key management and skilled scientific personnel; |
| · | third parties provide their services to us on a timely and effective basis; |
| · | we are able to maintain our required supply of raw materials, including krill oil; |
| · | we are able to find and retain a third-party to manufacture CaPre in compliance with cGMP; |
| · | we are able to successfully build a commercial organization, launch CaPre in the US, and compete in the US market; |
| · | we are able to secure distribution arrangements for CaPre, if it reaches commercialization; |
| · | we are able to manage our future growth effectively; |
| · | we are able to gain acceptance of CaPre in its markets and we are able to serve those markets; |
| · | our patent portfolio is sufficient and valid; |
| · | we are able to secure and defend our intellectual property rights and to avoid infringing upon the intellectual property rights of third parties; |
| · | we are able to take advantage of business opportunities in the pharmaceutical industry and receive strategic partner support; |
| · | we are able to continue as a going concern; |
| · | we are able to obtain additional capital and financing, as needed; |
| - 3 - |
| · | there is no significant increase in competition for CaPre from other companies in the pharmaceutical, medical food and natural health product industries; |
| · | CaPre would be viewed favorably by payers at launch and receive appropriate healthcare reimbursement; |
| · | market data and reports reviewed by us are accurate; |
| · | there are no adverse changes in relevant laws or regulations; and |
| · | we face no product liability lawsuits and other proceedings or any such matters, if they arise, are satisfactorily resolved. |
In addition, the forward-looking information in this annual report is subject to a number of known and unknown risks, uncertainties and other factors, including those described in this annual report under the heading “Item 3.D. Risk Factors”, many of which are beyond our control, that could cause our actual results and developments to differ materially from those that are disclosed in or implied by the forward-looking information, including, among others:
| · | risks related to timing and possible difficulties, delays or failures in our planned TRILOGY Phase 3 program for CaPre; |
| · | nonclinical and clinical trials may be more costly or take longer to complete than anticipated, and may never be initiated or completed, or may not generate results that warrant future development of CaPre; |
| · | CaPre may not prove to be as safe and effective or as potent as we currently believe; |
| · | our planned TRILOGY Phase 3 program may not produce positive results; |
| · | our anticipated studies and submissions to the FDA may not occur as currently anticipated, or at all; |
| · | the FDA could reject our 505(b)(2) regulatory pathway; |
| · | outcome study data from two of our competitors in high HTG patients may be negative, which could also negatively affect the market perception of CaPre; |
| · | we may encounter difficulties, delays or failures in obtaining regulatory approvals for the initiation of clinical trials or to market CaPre; |
| · | we may need to conduct additional future clinical trials for CaPre, the occurrence and success of which cannot be assured; |
| · | CaPre may have unknown side effects; |
| · | the FDA may refuse to approve CaPre, or place restrictions on our ability to commercialize CaPre; |
| · | CaPre could be subject to extensive post-market obligations and continued regulatory review, which may result in significant additional expense and affect sales, marketing and profitability; |
| · | we may fail to achieve our publicly announced milestones on time; |
| · | we may encounter difficulties in completing the development and commercialization of CaPre; |
| · | third parties we will rely upon to conduct our TRILOGY Phase 3 program for CaPre may not effectively fulfill their obligations to us, including complying with FDA requirements; |
| · | there may be difficulties, delays, or failures in obtaining health care reimbursements for CaPre; |
| · | recently enacted and future laws may increase the difficulty and cost for us to obtain marketing approval of and commercialize CaPre and affect the prices we can charge; |
| · | new laws, regulatory requirements, and the continuing efforts of governmental and third-party payors to contain or reduce the costs of healthcare through various means could adversely affect our business; |
| · | the market opportunity for, and demand and market acceptance of, CaPre may not be as strong as we anticipate; |
| · | third parties that we will rely upon to manufacture, supply and distribute CaPre may not effectively fulfill their obligations to us, including complying with FDA requirements; |
| · | there may not be an adequate supply of raw materials, including krill oil, in sufficient quantities and quality and to produce CaPre under cGMP standards; |
| · | Neptune still has some influence with respect to matters submitted to our shareholders for approval; |
| · | Neptune’s interest may not align with those of us or our other shareholders |
| - 4 - |
| · | we may not be able to meet applicable regulatory standards for the manufacture of CaPre or scale-up our manufacturing successfully; |
| · | we may not be able to produce clinical batches of CaPre in a timely manner or at all; |
| · | as a company, we have limited sales, marketing and distribution experience; |
| · | our patent applications may not result in issued patents, our issued patents may be circumvented or challenged and ultimately struck down, and we may not be able to successfully protect our trade secrets or other confidential proprietary information; |
| · | we may face claims of infringement of third party intellectual property and other proprietary rights; |
| · | we may face product liability claims and product recalls; |
| · | we face intense competition from other companies in the pharmaceutical, medical food and natural health product industries; |
| · | we have a history of negative operating cash flow and may never become profitable or be able to sustain profitability; |
| · | we have significant additional future capital needs and may not be able to raise additional financing required to fund further research and development, clinical studies, obtain regulatory approvals, build a commercial organization in the US, and meet ongoing capital requirements to continue our current operations on commercially acceptable terms or at all; |
| · | we may not be able to successfully compete in the US market with competitors who are larger and have more resources than we do; |
| · | we may acquire businesses or products or form strategic partnerships in the future that may not be successful; |
| · | we may be unable to secure development and/or distribution partnerships to support the development and commercialization of CaPre outside the US, provide development capital, or market access; |
| · | we rely on the retention of key management and skilled scientific personnel; and |
| · | general changes in economic and capital market conditions could adversely affect us. |
All of the forward-looking
information in this annual report is qualified by this cautionary statement. There can be no guarantee that the results or developments
that we anticipate will be realized or, even if substantially realized, that they will have the consequences or effects on our
business, financial condition or results of operations that we anticipate. As a result, you should not place undue reliance on
the forward-looking information. Except as required by applicable law, we do not undertake to update or amend any forward-looking
information, whether as a result of new information, future events or otherwise. All forward-looking information is made as of
the date of this annual report.
| - 5 - |
| Item 1. | Identity of Directors, Senior Management and Advisers |
Not applicable.
| Item 2. | Offer Statistics and Expected Timetable |
Not applicable.
| Item 3. | Key Information |
| A. | Selected Financial Data |
The following information should be read in conjunction with “Item 5. Operating and Financial Review and Prospects” and our audited financial statements and the related notes for our fiscal year ended March 31, 2018, which are prepared in accordance with IFRS as issued by the IASB and are included in this annual report. The selected financial information below includes financial information derived from our audited financial statements. Our historical results from any prior period are not necessarily indicative of results to be expected for any future period. The following table is a summary of our selected financial information in accordance with IFRS as issued by the IASB for each of our five most recently completed fiscal years.
| For the fiscal year ended | ||||||||||||||||||||
| March 31, 2018 | March 31, 2017 | February 29, 2016 | February 28, 2015 | February 28, 2014 | ||||||||||||||||
| Revenue from sales | $ nil | $ nil | $ nil | $ nil | $ | 501 | ||||||||||||||
| Loss from operating activities | $ | (19,696 | ) | $ | (11,210 | ) | $ | (9,612 | ) | $ | (12,395 | ) | $ | (10,800 | ) | |||||
| Net loss and total comprehensive loss | $ | (21,504 | ) | $ | (11,247 | ) | $ | (6,317 | ) | $ | (1,655 | ) | $ | (11,612 | ) | |||||
| Basic and diluted loss per share | $ | (1.23 | ) | $ | (1.01 | ) | $ | (0.59 | ) | $ | (0.16 | ) | $ | (1.38 | ) | |||||
| Total assets | $ | 22,959 | $ | 25,456 | $ | 28,517 | $ | 37,208 | $ | 45,632 | ||||||||||
| Total liabilities | $ | 14,735 | $ | 3,753 | $ | 1,297 | $ | 3,980 | $ | 12,352 | ||||||||||
| Share capital | $ |
73,338 |
$ | 66,576 | $ | 61,973 | $ | 61,628 | $ | 61,027 | ||||||||||
| Warrants and rights | $ | 715 |
$ | 453 | $ | — | $ | — | $ | 407 | ||||||||||
| Weighted average number of shares outstanding | 17,486,515 | 11,094,512 | 10,659,936 | 10,617,704 | 8,436,893 | |||||||||||||||
| Dividends declared per share | — | — | — | — | — | |||||||||||||||
| B. | Capitalization and Indebtedness |
Not applicable.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
| D. | Risk Factors |
Investing in our securities involves a high degree of risk due to, among other things, the nature of our business and the present stage of our development. Prospective and current investors should carefully consider the following risks and uncertainties, together with all other information in this annual report, as well as our financial statements included in this annual report and “Item 5. Operating and Financial Review and Prospects.” If any of these risks actually occur, our business, financial condition, prospects, results of operations or cash flow could be materially and adversely affected and you could lose all or a part of the value of your investment. Additional risks or uncertainties not currently known to us, or that we deem immaterial, may also negatively affect our business operations.”
General Risks Related to the Corporation
We may not be able to maintain our operations and advance our research and development of CaPre without additional funding.
We have incurred operating losses and negative cash flows from operations since our inception. To date, we have financed our operations through public offerings and private placements of securities, proceeds from exercises of warrants, rights and options, and receipt of research tax credits and research grant programs. Our cash and cash equivalents were $8.2 million as of March 31, 2018 and $9.8 million as of March 31, 2017.
| - 6 - |
Since our March 31, 2018 year end, the current assets have been increased by an incremental $10.0 million in approximate net proceeds from a May 2018 Canadian public financing, however, are projected to be less than needed to support our current liabilities as at that date when combined with the projected level of our expenses for the next twelve months, including the full initiation of clinical sites and ongoing enrollment of patients in, and the manufacturing of clinical materials for, our TRILOGY Phase 3 program for CaPre. Our positive working capital balance is expected to continue to decline until we raise additional funds. We will also require substantial additional funds to complete our TRILOGY Phase 3 program, obtain regulatory approvals and commercialize CaPre. In addition to completing nonclinical and clinical trials, we expect that additional time and capital will be required by us to file an NDA to obtain FDA approval for CaPre in the United States, to further scale up our manufacturing capabilities, and to complete marketing and other pre-commercialization activities. We will also most likely require additional capital to fund our daily operating needs. Based on a conservative estimate, we believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements through September 2018. To fully execute our business plan, we will need to raise the additional necessary capital primarily through additional securities offerings and strategic alliances in the near term. We currently have no other arranged sources of financing. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue our development or commercialization of CaPre or our other research and development initiatives. Delays or failures in our TRILOGY Phase 3 program for CaPre may affect our ability to complete strategic development and/or distribution partnerships to support the development and commercialization of CaPre. Additional funding from third parties may not be available on acceptable terms or at all to enable us to continue and complete our research and development of CaPre.
If we do not raise additional funds, we may not be able to realize our assets and discharge our liabilities in the normal course of business. As a result, there exists a material uncertainty that casts substantial doubt about our ability to continue as a going concern and, therefore, realize our assets and discharge our liabilities in the normal course of business. Our financial statements have been prepared on a going-concern basis, which assumes we will continue our operations in the foreseeable future and will be able to realize our assets and discharge our liabilities and commitments in the ordinary course of business. If we are unable to continue as a going concern, material write-downs to the carrying value of our assets, including intangible assets, could be required. If we fail to obtain additional financing, we may not be able to continue as a going concern.
We may never become profitable or be able to sustain profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. The likelihood of the success of our business plan must be considered in light of the problems, expenses, difficulties, complications and delays frequently encountered when developing and expanding early-stage businesses and the regulatory and competitive environment in which we operate. Biopharmaceutical product development is a highly speculative undertaking, involves a substantial degree of risk and is a capital- intensive business. We expect to incur expenses without any meaningful corresponding revenues unless and until we are able to obtain regulatory approval for and begin selling CaPre in significant quantities. We filed our investigational new drug application, or IND, for CaPre in late 2013, which allowed us to initiate clinical development in 2014 in the United States towards FDA approval for CaPre. To date, we have not generated any revenue from CaPre, and we may never be able to obtain regulatory approval for marketing CaPre in any indication. Even if we are able to commercialize CaPre, we may still not generate significant revenues or achieve profitability. Additionally, we may not be able to attain commercially viable cost of goods sold, and levels of insurance reimbursement for CaPre may not be commercially viable in all global markets. We incurred net losses for the fiscal year ended March 31, 2018 of $21.5 million, $11.2 million for the thirteen-month period ended March 31, 2017, and $6.3 million and $1.7 million for our fiscal years ended 2016 and 2015, respectively. As of March 31, 2018, we had an accumulated deficit of $72.4 million.
We expect that our expenses will increase significantly as we continue our Phase 3 clinical program for CaPre under the current indication and prepare to seek FDA approval for the commercial launch of CaPre. We also expect that our research and development expenses will continue to increase if we pursue FDA approval for CaPre for other indications. As a result, we expect to continue to incur substantial losses for the foreseeable future, and these losses may be increasing. We are uncertain about when or if we will be able to achieve or sustain profitability. If we fail to become and remain profitable, our ability to sustain our operations and to raise capital could be impaired and the price of our common shares could decline.
| - 7 - |
If outcome studies being conducted by two of our competitors testing the impact of OM3 on treating patients with high TGs are negative, there could also be an adverse impact for CaPre.
Two of our competitors are currently testing the effects of OM3 on patients with high TGs and taking statins concomitantly. These cardiovascular outcome studies are expected to report by the end of the third quarter of fiscal 2018 (the REDUCE-IT trial sponsored by Amarin) and in 2019 (the STRENGTH trial sponsored by AstraZeneca). If those studies show that OM3 therapeutic drugs effectively treat patients with high TGs and improve cardiovascular, morbidity and mortality outcomes, we believe that the potential to expand CaPre’s indication in the future to include the treatment of high TGs would be significantly advanced. Conversely, if outcome study data from one or both of those competitors is negative, or if one or both clinical trials fail to be completed, our potential target market for CaPre could be limited to patients with severe HTG (total global market was estimated by GOED Proprietary Research in 2015 to be approximately $2.3 billion) and our ability to realize greater market potential of CaPre could be harmed without conducting a successful outcomes trial with CaPre.
We will rely on third parties to conduct our TRILOGY Phase 3 program for CaPre.
We will rely on contract research organizations, or CROs, to monitor and manage data for our TRILOGY Phase 3 program for CaPre. While we will only control certain aspects of the CRO’s activities, we nevertheless are responsible for ensuring that our clinical trials are conducted in accordance with applicable protocols, and legal, regulatory and scientific standards, and our reliance on the CRO does not relieve us from those responsibilities. We and the CRO are required to comply with current good clinical practices, or cGCPs, which are regulations and guidelines enforced by the FDA, Health Canada and comparable foreign regulatory authorities for any products in clinical development.
The FDA enforces these cGCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or the CRO fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, Health Canada or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications for CaPre. Upon inspection, the FDA could determine that our clinical trials do not comply with cGCPs. In addition, our clinical trials must be conducted with products produced under current good manufacturing practice, or cGMP, regulations and require a large number of test subjects. If we or the CRO fail to comply with these regulations, we may have to repeat preclinical studies or clinical trials for CaPre, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
If our relationship with a CRO terminates, we may not be able to enter into arrangements with alternative CROs. If the CRO does not successfully carry out its duties or obligations or meet expected deadlines, if it needs to be replaced or if the quality or accuracy of the clinical data it obtains is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, we may have to extend, delay or terminate our preclinical studies or clinical trials, and we may not be able to obtain regulatory approval for or successfully commercialize CaPre.
The third parties that will help conduct our TRILOGY Phase 3 program for CaPre will not be our employees and, except for remedies available to us under our agreements with the CROs, we cannot control whether or not they devote sufficient time and resources to our preclinical, clinical and nonclinical programs. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other drug development activities, which could affect their performance on our behalf.
We rely on third parties to manufacture, produce and supply CaPre and we may be adversely affected if those third parties are unable or unwilling to fulfill their obligations, including complying with FDA requirements.
Producing pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Currently, while we do own our manufacturing and encapsulation equipment, we do not own or operate manufacturing facilities for the production of CaPre. Accordingly, we need to rely on one or more third party contract manufacturers to produce and supply our required drug product for our nonclinical research and clinical trials for CaPre.
Although we are currently working with CordenPharma at its Chenôve facility in Dijon, France to scale up our manufacturing processes for CaPre, doing so is a difficult and uncertain task, and there are risks associated with scaling to the level required for full commercialization, including, among others, pricing, cost overruns, potential problems with process scale up, process reproducibility, stability issues, lot consistency and timely availability of reagents or raw materials. Consequently, we may not be able to attain our targeted cost of goods sold for CaPre. Any of these challenges could delay completion of our clinical trials or commercial launch of CaPre, require bridging or repetition of studies or trials, increase development costs, delay approval of CaPre, impair our commercialization efforts, and increase our costs. We may have to delay or suspend the production of CaPre if a third-party manufacturer:
| · | becomes unavailable for any reason, including as a result of the failure to comply with cGMP regulations; |
| - 8 - |
| · | experiences manufacturing problems or other operational failures, such as equipment failures or unplanned facility shutdowns required to comply with cGMP or damage from any event, including fire, flood, earthquake, business restructuring or insolvency; or |
| · | fails or refuses to perform its contractual obligations under its agreement with us, such as failing or refusing to deliver the quantities of CaPre requested by us on a timely basis. |
If our third-party contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with cGMP regulations, we may be subject to sanctions, including fines, product recalls or seizures, injunctions, delays or suspensions of our clinical trials for CaPre, total or partial suspension of production of CaPre, civil penalties, withdrawals of previously granted regulatory approvals, and criminal prosecution. We do not currently have arrangements in place for redundant supply. If any one of our current contract manufacturers cannot perform as agreed, we may be required to replace that manufacturer. Although we believe that there are several potential alternative contract manufacturers who could manufacture CaPre, we may incur added costs and delays in identifying and qualifying any such replacement.
We depend on Neptune for certain administrative and accounting services.
Neptune has provided us in the past with certain shared back office services and functions, including corporate affairs, public company reporting, accounting, payroll, information technology, accounts payable, accounts receivable and shared premises. As of the date of this annual report, the corporate affairs, public company reporting, accounting, and accounts receivable services have not been renewed, and we are now incurring incremental costs to manage those functions independently ourselves. These additional costs are partially offset by reduced shared service fees, and we expect that these services will continue to be provided independently or through qualified third parties. If our arrangements with Neptune for the remaining services were to be terminated or not renewed, we may have to incur some additional costs to provide these services ourselves or to source them from another third party. We anticipate these operations to be fully independent of Neptune by the end of our 2019 fiscal year. However, there can be no assurances that this will fully materialize by such time. Currently, our arrangements with Neptune for the remaining services are on a month-to-month basis and can be terminated anytime by either Neptune or us.
We have historically had no marketing and sales organization and, as a company, no experience in marketing products. If we are unable to properly establish marketing and sales capabilities or enter into agreements with a strategic partner to market and sell CaPre, we may not be able to generate revenue.
We have historically had no sales, marketing or distribution capabilities and, as a company, we have also historically had no experience in marketing products. If CaPre or another of our future product candidates is approved for commercialization, unless we find a strategic partner to assist us with sales, marketing and distribution, we will be required to develop in-house marketing and sales force capability, which would require significant capital expenditures, management resources and time. Also, we would have to compete with other biotechnology and pharmaceutical companies to recruit, hire, train and retain marketing and sales personnel. We face competition in our search for strategic partners to assist us with sales, marketing and distribution, and we may not be able to establish or maintain any such arrangements. If we do find a strategic partner, any revenue we receive from CaPre would partly depend upon the efforts of that strategic partner, which may not be successful. We may have little or no control over the marketing and sales efforts by any strategic partner we find for CaPre and our revenue may be lower than if we had commercialized CaPre independently.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive pharmaceuticals industry largely depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. Competition for skilled personnel in our market is intense and competition for experienced scientists may limit our ability to hire and retain highly qualified personnel on acceptable terms. We are highly dependent on our management, scientific and medical personnel. Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice or, potentially, without any notice at all. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results or financial condition. Our success may also depend on our ability to attract, retain and motivate highly skilled junior, mid-level, and senior managers and scientific personnel. In addition, we do not maintain “key person” insurance policies on the lives of our executives or those of any of our other employees. Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high- quality candidates than what we can offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can develop and commercialize CaPre and any other future product candidates would be limited.
| - 9 - |
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our suppliers, third party manufacturers and other contractors and consultants could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self- insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to manufacture CaPre. Our ability to obtain supplies of CaPre could be disrupted if the operations of our manufacturers and suppliers are affected by a man-made or natural disaster or other business interruption.
Our prospects currently depend entirely on the success of CaPre, which is still in clinical development, and we may not be able to generate revenues from CaPre.
We have no prescription drug products that have been reviewed or approved by the FDA, Health Canada or any similar regulatory authority. Our only prescription drug candidate is CaPre, for which we have not yet filed an NDA, and for which we must conduct a TRILOGY Phase 3 program, undergo further development activities and seek and receive regulatory approval prior to commercial launch, which we do not anticipate will occur until 2021 at the earliest. We have invested significant effort and financial resources in researching and developing CaPre. Further development of CaPre will require substantial investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before we can generate any revenue from sales of CaPre, if it is ever approved for commercialization.
We do not have any other prescription drug candidates in development and so our business prospects currently depend entirely on the successful development, regulatory approval and commercialization of CaPre, which may never occur. Most prescription drug candidates never reach the clinical development stage and even those that do reach clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. If we are unable to successfully commercialize CaPre, we may never generate meaningful revenues. In addition, if CaPre reaches commercialization and there is low market demand for CaPre or the market for CaPre develops less rapidly than we anticipate, we may not have the ability to shift our resources to the development of alternative products.
If we encounter difficulties enrolling patients in our planned TRILOGY Phase 3 program, our development activities for CaPre could be delayed or otherwise adversely affected.
We may experience difficulties in patient enrollment in our clinical trials, including our planned TRILOGY Phase 3 program for CaPre, for a variety of reasons. Timely completion of our clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. The enrollment of patients depends on many factors, including:
| · | the number of clinical trials for other product candidates in the same therapeutic area that are currently in clinical development, and our ability to compete with those trials for patients and clinical trial sites; |
| · | patient eligibility criteria defined in the protocol; |
| · | the size of the patient population; |
| · | the risk that disease progression will result in death before the patient can enroll in clinical trials or before the completion of any clinical trials in which the patient is enrolled; |
| · | the proximity and availability of clinical trial sites for prospective patients; |
| · | the design of the trial; |
| · | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| · | our ability to obtain and maintain patient consents; and |
| · | the risk that patients enrolled in clinical trials will drop out of the trials before completion. |
Our planned TRILOGY Phase 3 program for CaPre may compete with other clinical trials for product candidates that are in the same therapeutic areas as CaPre. This competition could reduce the number and types of patients and qualified clinical investigators available to us, because some patients who might have opted to enroll in our TRILOGY Phase 3 program may instead opt to enroll in a trial being conducted by one of our competitors or a clinical trial site may not allow us to conduct our clinical program at that site if competing trials are already being conducted there. We may also encounter difficulties finding adequate clinical trial sites at which to conduct our TRILOGY Phase 3 program. Delays in patient enrollment may result in increased costs or may affect the timing or outcome of our planned TRILOGY Phase 3 program, which could impair or prevent its completion and adversely affect our ability to advance the development of CaPre.
| - 10 - |
We may not be able to obtain required regulatory approvals for CaPre.
We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA and, as a company, we have no experience in obtaining approval of any product candidates. The research, testing, manufacturing, labeling, packaging, storage, sale, marketing, pricing, export, import and distribution of prescription drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries and those regulations differ from country to country. We are not permitted to market CaPre in the United States until we receive approval of an NDA from the FDA and similar restrictions apply in other countries. In the United States, the FDA generally requires the completion of preclinical testing and clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are approved for commercialization. To date, we have not submitted an NDA for CaPre to the FDA or comparable applications to other regulatory authorities.
Our receipt of required regulatory approvals for CaPre is uncertain and subject to a number of risks, including:
| · | the FDA or comparable foreign regulatory authorities or independent institutional review boards, or IRBs, may disagree with the design or implementation of our clinical trials; |
| · | we may not be able to provide acceptable evidence of the safety and efficacy of CaPre; |
| · | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA or other regulatory agencies for marketing approval; |
| · | the dosing of CaPre in a particular clinical trial may not be at an optimal level; |
| · | patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to CaPre; |
| · | we may be unable to demonstrate that CaPre’s clinical and other benefits outweigh its safety risks; |
| · | the data collected from our clinical trials may not be sufficient to support the submission of an NDA for CaPre or to obtain regulatory approval for CaPre in the United States or elsewhere; |
| · | the FDA or comparable foreign regulatory authorities may not approve the manufacturing processes or facilities of third party manufacturers with which we contract for clinical and commercial supplies of CaPre; and |
| · | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
The FDA and other similar regulators have substantial discretion in the approval process and may refuse to accept our application or may decide that our data is insufficient for approval and require additional clinical trials, or preclinical or other studies for CaPre. If regulatory approval for CaPre is obtained in one jurisdiction that does not necessarily mean that CaPre will receive regulatory approval in all jurisdictions in which we seek approval. If we fail to obtain approval for CaPre in one or more jurisdictions, our ability to obtain approval in a different jurisdiction may be negatively affected.
Even if we receive regulatory approval for CaPre, it may just be for a limited indication.
If we obtain regulatory approval for CaPre, we will only be permitted to market it for the indication approved by the FDA, and any such approval may put limits on the indicated uses or promotional claims we may make for it, or otherwise not permit labeling that sufficiently differentiates CaPre from competitive products with comparable therapeutic profiles. For example, while our initial objective is to seek regulatory approval for the treatment of severe HTG, afterwards obtaining approval for CaPre to address mild to moderate HTG could greatly expand our potential market for CaPre. However, even if CaPre is approved for severe HTG, it may never be approved for the treatment of mild to moderate HTG. In addition, any approval we receive for CaPre could contain significant use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. If any regulatory approval for CaPre contains significant limits, we may not be able to obtain sufficient funding or generate meaningful revenue from CaPre or be able to continue developing, marketing or commercializing CaPre.
We may be unable to find successful strategic partnerships to develop and commercialize CaPre.
We intend to seek co-development, licensing and/or marketing partnership opportunities with third parties that we believe will complement or augment our development and commercialization efforts for CaPre. Entering into partnership relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing shareholders or disrupt our management and business. Entering into partnership relationships could also delay the development of CaPre and our other future product candidates if we become dependent upon a strategic partner and that strategic partner does not prioritize the development of CaPre relative to its other development activities. In addition, we face significant competition in seeking strategic partners and the negotiation process is time-consuming and complex. We may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for CaPre on our anticipated timeline, or at all, because CaPre may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view CaPre as having the requisite potential to demonstrate safety and efficacy. Even if we do enter into strategic partnerships, those partnerships may not achieve our objectives.
We are currently engaged in strategic partnership discussions with several pharmaceutical companies for the development and commercialization of CaPre. On November 20, 2017, we announced the signing of a non-exclusive non-binding term sheet with a leading China-based pharmaceutical company, and discussions with other parties are proceeding. Completion of any transaction is subject to negotiation and execution of a definitive agreement, which if signed would grant an exclusive license to commercialize CaPre in certain Asian countries, including China. Any signed preliminary agreements are preliminary and non-binding at this stage and the license, upfront payment, possible milestone payments and royalties contemplated by them will only become operative if definitive documents are executed. While the negotiation process remains ongoing with the view to reach a definitive agreement, the outcome at this point in time is uncertain and it is possible that no definitive agreement will be reached, or, if a definitive agreement is reached, that its terms and conditions may differ from those in the preliminary agreements. If we do enter into definitive documents, the near-term timing of the next steps in the advancement of our research and development of CaPre could be affected as the development of CaPre in those Asian countries may have to be pursued under a separate clinical program from our North American TRILOGY Phase 3 program.
| - 11 - |
We may be unable to develop alternative product candidates.
To date, we have not commercialized any prescription drug candidates and, other than CaPre, we do not have any compounds in clinical trials, nonclinical testing, lead optimization or lead identification stages. If we fail to obtain regulatory approval for and successfully commercialize CaPre as a treatment for severe HTG or any other indication, whether as a stand-alone therapy or in combination with other treatments, we would have to develop, acquire or license alternative product candidates or drug compounds to expand our product candidate pipeline beyond CaPre. In such a scenario, we may not be able to identify and develop or acquire product candidates that prove to be successful products, or to develop or acquire them on terms that are acceptable to us.
We may not be able to compete effectively against our competitors’ pharmaceutical products.
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to CaPre. It is probable that the number of companies seeking to develop products and therapies similar to CaPre will increase. Many of these and other existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. These companies may develop and introduce products and processes competitive with or superior to CaPre. In addition, other technologies or products may be developed that have an entirely different approach or means of accomplishing the intended purposes of CaPre, which might render our technology and CaPre non-competitive or obsolete.
Our competitors in the United States and globally include large, well-established pharmaceutical companies, specialty pharmaceutical sales and marketing companies, and specialized cardiovascular treatment companies. GlaxoSmithKline plc, which currently sells LOVAZA, a prescription-only OM3 fatty acid indicated for patients with severe HTG, was approved by FDA in 2004 and has been on the market in the United States since 2005. Multiple generic versions of LOVAZA are now available in the United States. Amarin launched its prescription-only OM3 drug VASCEPA in 2013, and reached a market share of approximately 20% by the end of 2015. In addition, EPANOVA (OM3-carboxylic acids) capsules, a free fatty acid form of OM3 (comprised of 55% EPA and 20% DHA), is FDA-approved for patients with severe HTG. Omtryg, another OM3 fatty acid composition developed by Trygg Pharma AS, received FDA approval for severe HTG. Neither EPANOVA nor Omtryg have yet been commercially launched, but could launch at any time. Other large companies with products competing indirectly with CaPre include AbbVie, Inc., which currently sells Tricor and Trilipix for the treatment of severe HTG, and Niaspan, which is primarily used to raise HDL-C but is also used to lower TGs. Generic versions of Tricor, Trilipix and Niaspan are also now available in the United States. In addition, we are aware of a number of other pharmaceutical companies that are developing products that, if approved and marketed, would compete with CaPre.
Even if it receives regulatory approval, CaPre may need to demonstrate compelling comparative advantages in efficacy, convenience, tolerability and safety to be commercially successful. Other competitive factors, including generic drug competition, could force us to lower prices or could result in reduced sales of CaPre. In addition, new products developed by others could emerge as competitors to CaPre. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
CaPre could face competition from products for which no prescription is required.
If it receives regulatory approval, CaPre will be a prescription-only OM3. Mixtures of OM3 fatty acids are naturally occurring substances in various foods, including fatty fish. OM3 fatty acids are also marketed by other companies as dietary supplements or natural health products. Dietary supplements may generally be marketed without a lengthy FDA premarket review and approval process and do not require a prescription. However, unlike prescription drug products, manufacturers of dietary supplements may not make therapeutic claims for their products; dietary supplements may be marketed with claims describing how the product affects the structure or function of the body without premarket approval, but may not expressly or implicitly represent that the dietary supplement will diagnose, cure, mitigate, treat, or prevent disease. We cannot be certain that physicians or consumers will view CaPre as superior to these alternatives or that physicians will be more likely to prescribe CaPre. If the price of CaPre is significantly higher than the prices of commercially available OM3 fatty acids marketed by other companies as dietary supplements or natural health products, physicians may recommend these commercial alternatives instead of CaPre or patients may elect on their own to take commercially available non-prescription OM3 fatty acids. Either of these outcomes could limit how we price CaPre and negatively affect our revenues.
Recent and future legal developments could make it more difficult and costly for us to obtain regulatory approvals for CaPre and negatively affect the prices we may charge.
In the United States and elsewhere, recent and proposed legal and regulatory changes to healthcare systems could prevent or delay our receipt of regulatory approval for CaPre, restrict or regulate our post-approval marketing activities, and adversely affect our ability to profitably sell CaPre. Proposals have also been made to expand post-approval requirements and to restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA’s regulations, guidance or interpretations will be changed, or what impact any such changes will have, if any, on our ability to obtain regulatory approvals for CaPre. Further, the Centers for Medicare and Medicaid Services, or CMS, frequently changes product descriptors, coverage policies, product and service codes, payment methodologies and reimbursement values. Also, increased scrutiny by the U.S. Congress of the FDA’s approval process could significantly delay or prevent our receipt of regulatory approval for CaPre and subject us to more stringent product labeling and post-marketing testing and other requirements.
| - 12 - |
In the United States, the Medicare Modernization Act, or the MMA, changed the way Medicare covers and pays for pharmaceutical products. The MMA expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, the MMA authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of the MMA and the expansion of federal coverage of drug products, we expect there will be additional pressure to contain and reduce healthcare costs. These healthcare cost reduction initiatives and other provisions of the MMA could decrease the coverage and price that we would receive for CaPre. While the MMA applies only to drug benefits for Medicare beneficiaries, private health insurance companies often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private health insurance companies.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (the Health Care Reform Law), has broadened access to health insurance, reduced or constrained the growth of healthcare spending, enhanced remedies against fraud and abuse, added new transparency requirements for the healthcare and health insurance industries, imposed new taxes and fees on the health industry and imposed additional health policy reforms. Provisions of the Health Care Reform Law affecting pharmaceutical companies include requirements to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “donut hole”, and to pay an annual non-tax deductible fee to the federal government based on each company’s market share of prior year total sales of branded products to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs and Department of Defense.
Despite initiatives to invalidate the Health Care Reform Law, the U.S. Supreme Court has upheld key aspects of it. Due to the results of the recent presidential election, the Health Care Reform Law may be significantly changed and we do not know whether any such changes could have significant negative financial impact on the development or potential profitability of CaPre. At this time, it remains unclear whether there will be any changes made to the Health Care Reform Law, whether to certain provisions or its entirety. The Health Care Reform Law or any replacement of it could continue to apply downward pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Additional federal healthcare reform measures could be adopted in the future limiting the amounts that federal and state governments will pay for healthcare products and services, which could negatively affect the value of CaPre and our ability to achieve profitability.
In Canada, most new patented drug prices are limited so that the cost of therapy is in the range of the cost of therapy for existing drugs sold in Canada used to treat the same disease. As a result:
| · | prices of moderate and substantial improvement drugs and breakthrough drugs are also restricted by a variety of tests; |
| · | existing patented drug prices cannot increase by more than the Canadian Consumer Price Index; and |
| · | the Canadian prices of patented medicines can never be the highest in the world. |
If CaPre receives regulatory approval in Canada, restrictions on the price we can charge there for CaPre could reduce the value of CaPre and our ability to generate revenue and achieve profitability.
In many jurisdictions outside the United States, a product candidate must be approved for health care reimbursement before it can be approved for sale. In some cases, the price that we intend to charge for CaPre will also be subject to approval. If we fail to comply with the regulatory requirements in our target international markets or to receive required marketing approvals, our potential market for CaPre will be reduced and our ability to realize the full market potential for CaPre will be harmed.
Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance. If there is not sufficient reimbursement for CaPre, it is less likely that it will be widely used.
Even if CaPre is approved for sale by the appropriate regulatory authorities, market acceptance and sales of CaPre will depend on reimbursement policies and may be affected by future healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will reimburse and establish payment levels. We cannot be certain that reimbursement will be available for CaPre. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize CaPre.
There may be significant delays in obtaining coverage and reimbursement for newly-approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or other regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution expenses. Interim reimbursement levels for new drugs, if applicable, may also be insufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of a drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and private payors for CaPre could have a material adverse effect on our operating results and our overall financial condition.
| - 13 - |
Even if we obtain FDA approval of CaPre, we may never obtain approval or commercialize it outside of the United States, which would limit our ability to realize CaPre’s full market potential.
In order to market CaPre outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and may require additional preclinical studies or clinical trials, which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of CaPre in those countries. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on the process for regulatory approval in other countries. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, our target market will be reduced and our ability to realize the full market potential of CaPre will be harmed.
If we or our third-party service providers fail to comply with healthcare laws and regulations or government price reporting laws, we could be subject to civil or criminal penalties.
In addition to the FDA’s restrictions on marketing pharmaceutical products, several other types of federal and state healthcare fraud and abuse laws restrict marketing practices in the pharmaceutical industry. These laws include the U.S. Anti-Kickback Statute, U.S. False Claims Act and similar state laws. The U.S. Anti-Kickback Statute prohibits, among other things, offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, or ordering any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. A person or entity does not need to have actual knowledge of the U.S. Anti-Kickback Statute or special intent to violate the law in order to have committed a violation. This statute has been interpreted broadly to apply to arrangements between pharmaceutical manufacturers and prescribers, dispensers, purchasers and formulary managers. The exemptions and safe harbors from prosecution are drawn narrowly and we may fail to meet all of the criteria for safe harbor protection from anti-kickback liability.
In addition, the Health Care Reform Law provides that the government may assert that a claim including items or services resulting from a violation of the U.S. Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the U.S. False Claims Act. Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. The “qui tam” provisions of the False Claims Act allow a private individual to bring civil actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government. These individuals, sometimes known as “relators” or, more commonly, as “whistleblowers”, may share in any amounts paid by the entity to the government in fines or settlement. The number of filings of qui tam actions has increased significantly in recent years, causing more healthcare companies to have to defend a case brought under the federal False Claim Act. If an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus attorneys’ fees and costs, and civil penalties of up to US$21,563 for each separate false claim. Certain administrative sanctions, up to and including exclusion of an entity from participation in the federal healthcare programs, may also ensue.
Additional laws and regulations include:
| · | the U.S. federal Health Insurance Portability and Accountability Act (HIPAA), as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), which created additional federal criminal statutes that prohibit, among other things, schemes to defraud healthcare programs and imposes requirements on certain types of people and entities relating to the privacy, security, and transmission of individually identifiable health information, and requires notification to affected individuals and regulatory authorities of breaches of security of individually identifiable health information; |
| · | the federal Physician Payment Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, to report annually to the CMS information related to payments and other transfers of value to physicians, other healthcare providers and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members, which is published in a searchable form on an annual basis; and |
| - 14 - |
| · | the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation. |
Over the past few years, a number of pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of alleged prohibited promotional and marketing activities, such as providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion that caused claims to be submitted to Medicaid for non-covered, off-label uses; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates. Most states also have statutes or regulations similar to the U.S. Anti-Kickback Statute and the U.S. False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment. Settlements of U.S. government litigation may include Corporate Integrity Agreements with commitments for monitoring, training, and reporting designed to prevent future violations.
Any action against us for an alleged or suspected violation of these laws could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with these laws and regulations may be costly to us in terms of money, time and resources. If we or any strategic partners, manufacturers or service providers fail to comply with these laws, we could be subject to enforcement actions, including:
| · | adverse regulatory inspection findings; |
| · | warning letters; |
| · | voluntary or mandatory product recalls or public notification or medical product safety alerts to healthcare professionals; |
| · | restrictions on, or prohibitions against, marketing our products; |
| · | restrictions on, or prohibitions against, importation or exportation of our products; |
| · | suspension of review or refusal to approve pending applications or supplements to approved applications; |
| · | exclusion from participation in government-funded healthcare programs; |
| · | exclusion from eligibility for the award of government contracts for our products; |
| · | suspension or withdrawal of product approvals; |
| · | product seizures; |
| · | injunctions; and |
| · | civil and criminal penalties and fines. |
We rely on third parties to conduct our clinical trials for CaPre.
We rely heavily on contract research organizations, or CROs, to monitor and manage data for our preclinical studies and clinical trials for CaPre. While we only control certain aspects of the CRO’s activities, we nevertheless are responsible for ensuring that our clinical trials are conducted in accordance with applicable protocols, legal, regulatory and scientific standards, and our reliance on the CRO does not relieve us from those responsibilities. We and the CRO are required to comply with cGCPs, which are regulations and guidelines enforced by the FDA, Health Canada and comparable foreign regulatory authorities for any products in clinical development.
The FDA enforces these cGCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or the CRO fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, Health Canada or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications for CaPre. Upon inspection, the FDA could determine that our clinical trials do not comply with cGCPs. In addition, our clinical trials must be conducted with products produced under cGMP regulations and require a large number of test subjects. If we or the CRO fail to comply with these regulations, we may have to repeat preclinical studies or clinical trials for CaPre, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
If our relationship with a CRO terminates, we may not be able to enter into arrangements with alternative CROs. If the CRO does not successfully carry out its duties or obligations or meet expected deadlines, if it needs to be replaced or if the quality or accuracy of the clinical data it obtains is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, we may have to extend, delay or terminate our preclinical studies or clinical trials, and we may not be able to obtain regulatory approval for or successfully commercialize CaPre.
| - 15 - |
The third parties conducting our preclinical studies and clinical trials at CROs will not be our employees and, except for remedies available to us under our agreements with the CROs, we cannot control whether or not they devote sufficient time and resources to our preclinical, clinical and nonclinical programs. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other drug development activities, which could affect their performance on our behalf.
The research, development and manufacture of CaPre involves using potentially hazardous materials.
Our research and development activities relating to CaPre involve the controlled use of potentially hazardous substances, including chemical materials such as acetone. Our manufacturers for CaPre will be subject to federal, provincial, state and local laws and regulations in Canada, the United States and in other jurisdictions governing laboratory procedures and the use, manufacture, storage, handling and disposal of medical and hazardous materials. Although we believe that our manufacturers’ procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we cannot completely eliminate the risk of contamination or injury resulting from medical or hazardous materials. If any such contamination or injury were to occur, we may incur liability or local, city, provincial, state or federal authorities may curtail the use of these materials and interrupt our business operations and the production of CaPre. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We do not have any insurance for liabilities arising from medical or hazardous materials. Complying with environmental, health and safety laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts relating to CaPre, which could harm our business, prospects, financial condition or results of operations. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of potentially hazardous materials. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These laws and regulations may make it more difficult for us to conduct our research, development or production activities relating to CaPre and if we fail to comply with them, we could have substantial fines, penalties or other sanctions imposed against us.
Interruptions of our supply of CaPre could disrupt our planned TRILOGY Phase 3 program and, if CaPre reaches commercialization, impair any future revenue streams.
We will require much larger amounts of CaPre for purposes of our planned TRILOGY Phase 3 program and potential commercialization than we have in the past. Supply interruptions for CaPre could occur and our inventory of CaPre may not always be sufficient due to a number of factors, including:
| · | failure to have a third-party supply chain partner’s process validated in a timely manner; |
| · | shortages of required raw materials, such as krill oil, and the packaging components required by our manufacturers; |
| · | changes in our sources for manufacturing or packaging; |
| · | failure to timely locate and obtain replacement manufacturers, as needed; and |
| · | conditions affecting the cost and availability of raw materials. |
We are also in the process of scaling-up our production of CaPre and CaPre may not be of comparable quality when produced in large 100 kilogram batches. If we experience interruptions in the production of CaPre, our ability to complete our planned TRILOGY Phase 3 program could be interrupted. If CaPre receives regulatory approval, interruptions in the production of CaPre or insufficient inventory levels of CaPre could have a material adverse effect on our results of operations.
If product liability lawsuits are brought against us, we may incur substantial liabilities and be required to cease the sale, marketing and distribution of CaPre.
We face a potential risk of product liability associated with any future commercialization of CaPre or any other future product candidate we develop. For example, we may be sued if CaPre allegedly causes injury. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under U.S. state or Canadian provincial or other foreign consumer protection legislation. If we cannot successfully defend against product liability claims, we may incur substantial liabilities or be required to cease the sale, marketing and distribution of CaPre. Even successful defense against product liability claims would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for CaPre or any future products that we may develop; |
| - 16 - |
| · | injury to our reputation; |
| · | costs to defend the related litigation; |
| · | a diversion of management’s time and our resources; |
| · | substantial monetary awards to consumers, trial participants or patients; |
| · | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | loss of revenue; |
| · | an inability to commercialize CaPre; and |
| · | a decline in the price of our common shares. |
If we are unable to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims, the commercialization of CaPre or any other product candidates we develop could be hindered or prevented. We currently carry product liability insurance, shared with Neptune, in the amount of $10.0 million in the aggregate. Any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. In the event of a successful product liability claim against us, we may have to pay from our own resources any amounts awarded by a court or negotiated in a settlement that exceed coverage limitations or that is not covered by our insurance, and we may not have, or be able to obtain, sufficient funds to pay such amounts.
We may not achieve our publicly announced milestones on time, or at all.
From time to time, we may publicly announce the timing of certain events we expect to occur, such as the anticipated timing of results from our clinical trials. These statements are forward-looking and are based on the best estimate of management at the time relating to the occurrence of the events. However, the actual timing of these events may differ from what has been publicly disclosed. The timing of events such as completion of a clinical trial, discovery of a new product candidate, filing of an application to obtain regulatory approval, beginning of commercialization of products, or announcement of additional clinical trials for a product candidate may ultimately vary from what is publicly disclosed. For example, we cannot provide assurances that we will conduct our planned Phase 3 clinical trial for CaPre, that we will make regulatory submissions or receive regulatory approvals as planned, or that we will be able to adhere to plans for the scale-up of manufacturing and launch of CaPre. These variations in timing may occur as a result of different events, including the nature of the results obtained during a clinical trial or during a research phase, problems with a supplier or a distribution partner or any other event having the effect of delaying the publicly announced timeline. We undertake no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously-announced milestones could have a material adverse effect on our business, financial condition or operating results and the trading price of our common shares.
We may be subject to foreign exchange rate fluctuations
Our reporting currency is the Canadian dollar. However, many of our expenses, such as CaPre’s chief manufacturing organization’s production activities and certain CRO arrangements for our planned TRILOGY Phase 3 program, currently are and/or are expected to be, denominated in foreign currencies, including European euros and U.S. dollars. As we currently complete financings in both Canadian and U.S. dollars, both currencies are maintained and used to make its needed payments in the applicable currency. Though we plan to implement measures designed to reduce our foreign exchange rate exposure, the U.S. dollar/Canadian dollar and European euro/Canadian dollar exchange rates have fluctuated significantly in the recent past and may continue to do so, which could have a material adverse effect on our business, financial position and results of operations.
In the past, Neptune supplied us with the krill oil needed to produce CaPre for our clinical programs, including the krill oil projected to be needed for our TRILOGY Phase 3 program, and we are now evaluating alternative suppliers.
We have sourced all of our krill oil from Neptune in the past to produce CaPre. We have sufficient krill oil inventories that we anticipate will be required to complete our TRILOGY Phase 3 program. However, in light of Neptune’s announcement of its plan to discontinue krill oil production and the sale of its krill oil inventory to Aker, we are evaluating alternative suppliers of krill oil. While we believe that alternative supplies of krill oil that can meet our specifications will be readily available, any alternative supply of krill oil may not be of comparable quality or cost to that provided by Neptune, which could negatively affect CaPre. Our reliance on third-party suppliers for krill oil exposes us to risks such as potential fluctuations in supply and reduced control over our production costs and delivery schedules for CaPre.
| - 17 - |
CaPre may cause or be perceived to cause undesirable side effects or have other properties that could delay or prevent its regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Many of the patients that we expect to enroll in our planned clinical trial may have pre-existing disorders. While such disorders may lead to serious adverse events during the clinical trial that may be found to be unrelated to CaPre, such events may create a negative safety perception and adversely impact market acceptance of CaPre following any approval.
If unacceptable side effects arise during the clinical trials for CaPre, we, the FDA or comparable foreign regulatory authorities, the Institutional Review Boards, or IRBs, or independent ethics committees at the institutions in which our studies are conducted, could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Side effects, whether treatment-related or not, could also affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. Inadequate training in recognizing or managing the potential side effects of CaPre could result in patient injury. Any of these occurrences may harm our business, financial condition and prospects significantly.
Moreover, clinical trials of CaPre are conducted in carefully defined sets of patients who have agreed to enter into clinical trials. Consequently, it is possible that our clinical trials, or those of any future collaborator or third party researcher, may indicate an apparent positive effect of CaPre that is greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of a product candidate, we, or others, discover that the product is less effective than previously believed or causes undesirable side effects that were not previously identified during the clinical trial phase, any of the following adverse events could occur:
| · | regulatory authorities may withdraw their approval of the product or seize the product; |
| · | we, or any future collaborators or third party researcher, may need to recall the product, or be required to change the way the product is administered or conduct additional clinical trials; |
| · | restrictions may be imposed on the marketing of, or the manufacturing processes for CaPre; |
| · | we may be subject to fines, injunctions or the imposition of civil or criminal penalties; |
| · | regulatory authorities may require the addition of labeling statements; |
| · | we, or any future collaborators, may be required to issue a communication outlining the risks of the previously unidentified side effects for distribution to patients; |
| · | we, or any future collaborators, could be sued and held liable for harm caused to patients; |
| · | CaPre may become less competitive; and |
| · | our reputation may suffer. |
Any of these events could harm our business and operations and could negatively impact our share price.
Risks Related to Intellectual Property
In addition to our own patents, CaPre is covered by patents that are sublicensed to us by Neptune.
In addition to our proprietary patent applications, we have a license under the License Agreement to use certain intellectual property of Neptune to develop and commercialize CaPre and our novel and active pharmaceutical ingredients, or APIs, for use in pharmaceutical and medical food applications in the cardiovascular field.
Moreover, the intellectual property which was licensed to us has recently been acquired by Aker. Aker has granted to Neptune the right to sublicense to us certain intellectual property as necessary to allow us to maintain its license grant under the License Agreement. Accordingly, the license granted to us under the License Agreement remains in full force.
Disputes may arise between us and Neptune or Aker regarding the intellectual property that is subject to the License Agreement, including with respect to:
| · | the scope of rights granted under the License Agreement and other interpretation-related issues; and |
| · | our right to sublicense patent and other rights to third parties under collaborative development relationships. |
| - 18 - |
If our sublicense with Neptune is terminated due to a breach by us of its terms (or should the License Agreement otherwise terminate, and we are unable to enter into a direct license agreement with Aker), we may not be able to manufacture and market CaPre prior to these patents expiration in 2022. This could delay our launch by 6 to 12 months, which would have a material adverse effect on our business and financial condition.
It is difficult and costly to protect our intellectual property rights.
The success of our business will largely depend on our ability to:
| · | obtain and maintain patents, trade secret protection and operate without infringing the intellectual proprietary rights of third parties; |
| · | successfully defend our patents, including patents licensed to us by Neptune, against third-party challenges; and |
| · | successfully enforce our patents against third party competitors. |
Our patents and/or proprietary technologies could be circumvented through the adoption of competitive, though non-infringing, processes or products. The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws may diminish the value of our intellectual property. We cannot predict the breadth of claims that may be allowable or enforceable in our patents, including the patents licensed to us by Neptune.
We face risks that:
| · | our rights under our Canadian, U.S. or foreign patents or other patents that Neptune or other third parties license to us could be curtailed; |
| · | we may not be the first inventor of inventions covered by our issued patents or pending applications or be the first to file patent applications for those inventions; |
| · | our pending or future patent applications may not be issued with the breadth of claim coverage sought by us, or be issued at all; |
| · | our competitors could independently develop or patent technologies that are substantially equivalent or superior to our technologies; |
| · | our trade secrets could be learned independently by our competitors; |
| · | the steps we take to protect our intellectual property may not be adequate; and |
| · | effective patent, trademark, copyright and trade secret protection may be unavailable, limited or not sought by us in some foreign countries. |
Further, patents have a limited lifespan. In the United States, a patent generally expires 20 years after it is filed (or 20 years after the filing date of the first non-provisional U.S. patent application to which it claims priority). While extensions may be available, the life of a patent, and the protection it affords, is limited. Without patent protection for CaPre or any other of our future product candidates, we may be open to competition from generic versions of CaPre or our other future product candidates. Further, the extensive period of time between patent filing and regulatory approval for a product candidate limits the time during which we can market that product candidate under patent protection. Patents owned by third parties could have priority over patent applications filed or in-licensed by us, or we or our licensors could become involved in interference, opposition or invalidity proceedings before U.S., Canadian or foreign patent offices. The cost of defending and enforcing our patent rights against infringement charges by other patent holders may be significant and could limit our operations.
| - 19 - |
CaPre may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development and commercialization efforts.
Our success depends in part on avoiding infringement of the proprietary technologies of others. The pharmaceutical industry has been characterized by frequent litigation regarding patent and other intellectual property rights. Identification of third party patent rights that may be relevant to our proprietary or licensed technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. Additionally, because patent applications are maintained in secrecy until the application is published, we may be unaware of third-party patents that may be infringed by our development and commercialization of CaPre or any other future product candidate. There may be certain issued patents and patent applications claiming subject matter that we may be required to license in order to research, develop or commercialize CaPre, and any such patents and patent applications may not be available to license on commercially reasonable terms, or at all. If claims of patent infringement are asserted by third parties against us, they could be time-consuming and may:
| · | result in costly litigation; |
| · | divert the time and attention of our technical personnel and management; |
| · | delay our clinical trials for CaPre; |
| · | prevent us from commercializing CaPre until the asserted patent expires or is held finally invalid or not infringed in court; |
| · | require us to cease or to modify our use of the technology and/or develop non-infringing technology; or |
| · | require us to enter into royalty or licensing agreements. |
Others may hold proprietary rights that could prevent CaPre from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin commercial activities relating to CaPre or our processes could subject us to potential liability for damages and require us to obtain a license to continue to manufacture or market CaPre or any other future prescription drug candidates. We might not prevail in any such actions or if any license is required under any of these patents it may not be available on commercially acceptable terms, if at all.
Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. We could be forced to redesign CaPre or any other future product candidates or processes to avoid infringement.
In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
A number of companies, including several major pharmaceutical companies, have conducted research on pharmaceutical uses of OM3 fatty acids, which has resulted in the filing of many patent applications related to this research. We are aware of third-party U.S., Canadian or other foreign patents that contain broad claims related to methods of using these general types of compounds, which may be construed to include potential uses of CaPre. If we were to challenge the validity of these or any other issued U.S., Canadian or other foreign patents in court, we would need to overcome a statutory presumption of validity that attaches to every U.S. and Canadian patent. This means that, in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the other party’s patent’s claims. If we were to challenge the validity of any issued U.S. patent in an administrative trial before the Patent Trial and Appeal Board in the United States Patent and Trademark Office, or USPTO, we would have to prove that the claims are unpatentable by a preponderance of the evidence. If there are disputes over our intellectual property rights, a jury and/or court may not find in our favor on questions of infringement, validity or enforceability.
If we do not protect our trademark for CaPre, we may not be able to build name recognition in our markets of interest.
We have trademarked CaPre. Our trademark may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to this trademark or may be forced to stop using this name, which we need for name recognition by potential strategic partners and customers. If we are unable to establish name recognition based on our trademark, we may not be able to compete effectively, and our business may be adversely affected.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time- consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. If we or our licensors were to initiate legal proceedings against a third party to enforce a patent covering CaPre or our technology, the defendant could counterclaim that our or our licensor’s patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements; for example, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we or our licensors and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on CaPre or certain aspects of our platform technology. Such a loss of patent protection could have a material adverse impact on our business. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without legally infringing our patents or other intellectual property rights.
| - 20 - |
In addition, in an infringement proceeding, a court may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business.
Interference proceedings provoked by third parties or brought by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could result in a loss of our current patent rights and could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, or at all. Litigation or interference proceedings may result in a decision adverse to our interests and, even if we are successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common shares.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect CaPre and any of our other future product candidates.
Numerous recent changes to the patent laws and proposed changes to the rules of the various Patent Offices around the world may have a significant impact on our ability to protect our technology and enforce our intellectual property rights. These changes may lead to increasing uncertainty with regard to the scope and value of our issued patents and to our ability to obtain patents in the future.
Once granted, patents may remain open to opposition, interference, re-examination, post-grant review, inter partes review, nullification derivation and opposition proceedings in court or before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against the initial grant. In the course of any such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims attacked, or may lose the allowed or granted claims altogether. Depending on decisions by authorities in various jurisdictions, the laws and regulations governing patents could change in unpredictable ways that may weaken our and our licensors’ ability to obtain new patents or to enforce existing patents we and our licensors or partners may obtain in the future.
We may not be able to protect our intellectual property rights throughout the world.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Risks Relating to Our Common Shares
The price of our common shares may be volatile.
Market prices for securities in general, and specifically those of development stage pharmaceutical companies, tend to fluctuate. Factors such as the announcement to the public or in various scientific or industry forums of technological innovations; new commercial products; patents, exclusive rights obtained by us or others; disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; the commencement, enrollment or results of future clinical trials it may conduct, or changes in the development status of its product candidates; results or delays of pre-clinical and clinical studies by us or others; any delay in its regulatory filings for its product candidates and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings; a change of regulations; additions or departures of key personnel; overall performance of the equity markets; general political and economic conditions; publications; failure to meet the estimates and projections of the investment community or that it may otherwise provide to the public; research reports or positive or negative recommendations or withdrawal of research coverage by securities analysts; actual or anticipated variations in quarterly operating results; announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; public concerns over the risks of pharmaceutical products and dietary supplements; unanticipated serious safety concerns related to the use of CaPre; the ability to finance, future sales of securities by us or our shareholders; and many other factors, many of which are beyond our control, could have considerable effects on the price of our securities. There can be no assurance that the market price of our common shares will not experience significant fluctuations in the future. As a result of any of these factors, the market price of our securities at any given point in time may not accurately reflect our value or the value of our securities.
| - 21 - |
In addition, the stock market in general, and pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common shares, regardless of our actual operating performance. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which would harm our business, operating results or financial condition.
Forward-Looking Statements may prove to be inaccurate.
Investors should not place undue reliance on forward-looking statements. By their nature, forward-looking statements involve numerous assumptions, known and unknown risks and uncertainties, of both general and specific nature, that could cause actual results to differ materially from those suggested by the forward-looking statements or contribute to the possibility that predictions, forecasts or projections will prove to be materially inaccurate. Additional information on the risks, assumptions and uncertainties can be found in this Prospectus under the heading “Forward-Looking Information”.
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of its shareholders will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of the shareholders. The incurrence of indebtedness would result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on its ability to incur additional debt, limitations on its ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact its ability to conduct its business. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, it may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
There can be no assurance that an active market for our common shares will be sustained.
There can be no assurance that an active market for our common shares will be sustained. Holders of common shares may be unable to sell their investments on satisfactory terms. As a result of any risk factor discussed herein, the market price of our common shares at any given point in time may not accurately reflect our long-term value. Furthermore, responding to these risk factors could result in substantial costs and divert management’s attention and resources. Substantial and potentially permanent declines in the value of our common shares may result and adversely affect the liquidity of the market for our common shares.
Other factors unrelated to our performance that may have an effect on the price and liquidity of our common shares include: extent of analyst coverage; lessening in trading volume and general market interest in our common shares; the size of our public float; and any event resulting in a delisting of our common shares.
A large number of common shares may be issued and subsequently sold upon the exercise of existing warrants. The sale or availability for sale of existing warrants or other securities convertible in common shares may depress the price of our common shares.
To the extent that holders of warrants sell common shares issued upon the exercise of warrants, the market price of our common shares may decrease due to the additional selling pressure in the market. The risk of dilution from issuances of common shares underlying existing warrants may cause shareholders to sell their common shares, which could further contribute to any decline in our common share market price.
Any downward pressure on the price of our common shares caused by the sale of common shares issued upon the exercise of existing warrants could encourage short sales by third parties. In a short sale, a prospective seller borrows common shares from a shareholder or broker and sells the borrowed common shares. The prospective seller anticipates that the common share price will decline, at which time the seller can purchase common shares at a lower price for delivery back to the lender. The seller profits when the common share price declines because it is purchasing common shares at a price lower than the sale price of the borrowed common shares. Such short sales of common shares could place downward pressure on the price of our common shares by increasing the number of common shares being sold, which could lead to a decline in the market price of our common shares.
| - 22 - |
We do not currently intend to pay any cash dividends on our common shares in the foreseeable future.
We have never paid any cash dividends on our common shares and we do not anticipate paying any cash dividends on our common shares in the foreseeable future because, among other reasons, we currently intend to retain any future earnings to finance our business. The future payment of cash dividends will be dependent on factors such as cash on hand and achieving profitability, the financial requirements to fund growth, our general financial condition and other factors our board of directors may consider appropriate in the circumstances. Until we pay cash dividends, which we may never do, our shareholders will not be able to receive a return on their common shares unless they sell them. See “Dividend Policy”.
If we fail to meet applicable listing requirements, the NASDAQ Stock Market or the TSXV may delist our common shares from trading, in which case the liquidity and market price of our common shares could decline.
Our common stock is currently listed on the NASDAQ Stock Market and the TSXV, but we cannot assure you that our securities will continue to be listed on the NASDAQ Stock Market and the TSXV in the future. In the past, we have received notices from the NASDAQ Stock Market that we have not been in compliance with its continued listing standards, and we have taken responsive actions and regained compliance. If we fail to comply with listing standards and the NASDAQ Stock Market or TSXV delists our common shares, we and our shareholders could face significant material adverse consequences, including:
| · | a limited availability of market quotations for our common shares; |
| · | reduced liquidity for our common shares; |
| · | a determination that our common shares are “penny stock”, which would require brokers trading in our common shares to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our common shares; |
| · | a limited amount of news about us and analyst coverage of us; and |
| · | a decreased ability for us to issue additional equity securities or obtain additional equity or debt financing in the future. |
We may pursue opportunities or transactions that adversely affect our business and financial condition.
Our management, in the ordinary course of our business, regularly explores potential strategic opportunities and transactions. These opportunities and transactions may include strategic joint venture relationships, significant debt or equity investments in Acasti by third parties, the acquisition or disposition of material assets, the licensing, acquisition or disposition of material intellectual property, the development of new product lines or new applications for our existing products, significant distribution arrangements, the sale of our common shares and other similar opportunities and transactions. The public announcement of any of these or similar strategic opportunities or transactions might have a significant effect on the price of our common shares. Our policy is to not publicly disclose the pursuit of a potential strategic opportunity or transaction unless it is required to do so by applicable law, including applicable securities laws relating to continuous disclosure obligations. There can be no assurance that investors who buy or sell securities are doing so at a time when we are not pursuing a particular strategic opportunity or transaction that, when announced, would have a significant effect on the price of our common shares.
In addition, any such future corporate development may be accompanied by certain risks, including exposure to unknown liabilities of the strategic opportunities and transactions, higher than anticipated transaction costs and expenses, the difficulty and expense of integrating operations and personnel of any acquired companies, disruption of our ongoing business, diversion of management’s time and attention, and possible dilution to shareholders. We may not be able to successfully overcome these risks and other problems associated with any future acquisitions and this may adversely affect our business and financial condition.
As a foreign private issuer, we are subject to different U.S. securities laws and regulations than a domestic U.S. issuer, which may limit the information publicly available to our U.S. shareholders.
We are a foreign private issuer under applicable U.S. federal securities laws, and therefore, we are not required to comply with all the periodic disclosure and current reporting requirements of the U.S. Securities and Exchange Act of 1934, or the Exchange Act. As a result, we do not file the same reports that a U.S. domestic issuer would file with the SEC, although we are required to file with or furnish to the SEC the continuous disclosure documents that we are required to file in Canada under Canadian securities laws. In addition, our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions of Section 16 of the Exchange Act. Therefore, our shareholders may not know on as timely a basis when our officers, directors and principal shareholders purchase or sell common shares as the reporting periods under the corresponding Canadian insider reporting requirements are longer. In addition, as a foreign private issuer, we are exempt from the proxy rules under the Exchange Act.
| - 23 - |
We cannot be certain that we will qualify as a foreign private issuer for our next fiscal year. If we no longer qualify as a foreign private issuer, we will no longer be exempt from the more stringent disclosure requirements applicable to U.S. companies.
As an “emerging growth company”, we are exempt from the requirement to comply with the auditor attestation requirements of the Sarbanes-Oxley Act.
We are an “emerging growth company”, as defined in the U.S. Jumpstart Our Business Start-ups Act, and we use the exemption provided to emerging growth companies from the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002. Therefore, our internal controls over financial reporting will not receive the level of review provided by the process relating to the auditor attestation included in annual reports of issuers that are not using an exemption. In addition, we cannot predict if investors will find our common shares less attractive because we rely on this exemption. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and trading price for our common shares may be negatively affected.
U.S. investors may be unable to enforce certain judgments.
We are a company existing under the Business Corporations Act (Québec). Some of our directors and officers are residents of Canada, and substantially all of our assets are located outside the United States. As a result, it may be difficult to effect service within the United States upon us or upon some of our directors and officers. Execution by U.S. courts of any judgment obtained against us or any of our directors or officers in U.S. courts may be limited to assets located in the United States. It may also be difficult for holders of securities who reside in the United States to realize in the United States upon judgments of U.S. courts predicated upon civil liability of us and our directors and executive officers under the U.S. federal securities laws. There may be doubt as to the enforceability in Canada against non-U.S. entities or their controlling persons, directors and officers who are not residents of the United States, in original actions or in actions for enforcement of judgments of U.S. courts, of liabilities predicated solely upon U.S. federal or state securities laws.
| Item 4. | Information on the Company |
| A. | History and Development of the Company |
Acasti was incorporated on February 1, 2002 under Part 1A of the Companies Act (Québec) under the name “9113-0310 Québec Inc.”. On February 14, 2011, the Business Corporations Act (Québec) (“QBCA”) came into effect and replaced the Companies Act (Québec). Acasti is now governed by the QBCA. On August 7, 2008, pursuant to a Certificate of Amendment, the Corporation changed its name to “Acasti Pharma Inc.”, its share capital description, the provisions regarding the restriction on securities transfers and the borrowing powers of the Corporation. On November 7, 2008, pursuant to a Certificate of Amendment, the Corporation changed the provisions regarding its borrowing powers. The Corporation became a reporting issuer in the Province of Québec on November 17, 2008.
Acasti’s head and registered office is located at 545 Promenade du Centropolis, Suite 100, Laval, Québec H7T 0A3. The Corporation currently employs 24 full-time employees with the majority working out of the Corporation’s headquarters in Laval and its laboratory in Sherbrooke. The Corporation’s website address is http://www.acastipharma.com. The Corporation does not incorporate the information on or accessible through its website into this Prospectus, and you should not consider any information on, or that can be accessed through, its website as part of this Prospectus.
Intercorporate Relationships
The Corporation has no subsidiaries. As of the date of this annual report, Neptune Technologies & Bioressources Inc. (“Neptune”) owns 5,064,694 Common Shares, representing approximately 13.8% of the issued and outstanding Common Shares.
| B. | Our Business |
We are a biopharmaceutical innovator focused on the research, development and commercialization of prescription drugs using omega-3, or OM3, fatty acids derived from krill oil. OM3 fatty acids have extensive clinical evidence of safety and efficacy in lowering triglycerides, or TGs, in patients with hypertriglyceridemia, or HTG. Our lead product candidate is CaPre, an OM3 phospholipid therapeutic, which we are developing initially for the treatment of severe HTG, a condition characterized by very high or severe levels of TGs in the bloodstream (≥ 500 mg/dL). In accordance with a study published in 2009 in the Archives of Internal Medicine by Ford et al., it is estimated that three to four million people in the United States have severe HTG. In the market research commissioned by us, physicians interviewed indicated a significant unmet medical need exists for an effective, safe and well-absorbing OM3 therapeutic that can also demonstrate a positive impact on the major blood lipids associated with cardiovascular, or CV, disease risk. We believe that CaPre will address this unmet medical need, if our Phase 3 results reproduce what we observed in our Phase 2 data. We initiated TRILOGY, our Phase 3 clinical program in North America during the second half of 2017 and started clinical site activation as planned at the end of 2017. As of the date of this annual report, patients are being actively enrolled and randomized for both studies. We also believe the potential exists to expand CaPre’s initial indication to the roughly 36 million patients with high TGs (blood levels between 200 – 499 mg/dL), although at least one additional clinical trial would likely be required to expand CaPre’s indications to this segment. We may also seek to identify new potential indications for CaPre that may be appropriate for future studies and pipeline expansion. In addition, we may also seek to in-license other cardiometabolic drug candidates for drug development and commercialization.
| - 24 - |
In four clinical trials conducted to date, we saw the following beneficial effects with CaPre, and we are seeking to demonstrate similar safety and efficacy in our planned TRILOGY Phase 3 program:
| · | significant reduction of TGs and non-high density lipoprotein cholesterol (non-HDL-C) levels in the blood of patients with mild to severe HTG; |
| · | no deleterious effect on low-density lipoprotein cholesterol (LDL-C), or “bad” cholesterol, with the potential to reduce LDL-C; |
| · | potential to increase high-density lipoprotein cholesterol (HDL-C), or “good” cholesterol; |
| · | good bioavailability (absorption by the body), even under fasting conditions; |
| · | no significant food effect when taken with either low-fat or high-fat meals; and |
| · | an overall safety profile similar to that demonstrated by currently marketed OM3s. |
We believe that these features could set CaPre apart from current FDA-approved OM3 treatment options, and could give us a significant clinical and marketing advantage.
About Hypertriglyceridemia
According to the American Heart Association Scientific Statement on Triglycerides and Cardiovascular Disease from 2011, TG levels provide important information as a marker associated with the risk for heart disease and stroke, especially when an individual also has low levels of HDL-C and elevated levels of LDL-C. HTG can be caused by both genetic and environmental factors, including obesity, sedentary lifestyle and high-calorie diets. HTG is also associated with comorbid conditions such as chronic renal failure, pancreatitis, nephrotic syndrome, and diabetes. Multiple epidemiological, clinical, genetic studies suggest that patients with elevated TG levels (≥ 200 mg/dL) are at a greater risk of coronary artery disease, or CAD, and pancreatitis, a life-threatening condition, as compared to those with normal TG levels. The genes regulating TGs and LDL-C are equally strong predictors of CAD, but HDL-C is not. Other studies suggest that lowering and managing TG levels may reduce these risks. In addition, the Japan EPA Lipid Intervention Study, or JELIS, demonstrated the long-term benefit of an OM3 eicosapentaenoic acid, or EPA, in preventing major coronary events in hypercholesterolemic patients receiving statin treatment. JELIS found a 19% relative risk reduction in major coronary events in patients with relatively normal TGs but a more pronounced 53% reduction in the subgroup with TGs > 150mg/dL and HDL-C < 40mg/dL. Recently published meta-analyses by Alexander et al. (Mayo Clinic Proceedings, 2017) and Maki et al. (Journal of Clinical Lipidology, 2016) suggest that EPA and docosahexaenoic acid, or DHA, may be associated with reducing coronary heart disease risk to a greater extent in populations with elevated TG levels, and that drugs lowering TG and TG-rich lipoproteins may reduce cardiovascular event risk in patients with elevated TG levels, particularly if associated with low HDL-C.
About CaPre
CaPre is a highly purified, proprietary krill oil-derived mixture containing polyunsaturated fatty acids, or PUFAs, primarily composed of OM3 fatty acids, principally eicosapentaenoic acid, or EPA, and docosahexaenoic acid, or DHA, present as a combination of phospholipid esters and free fatty acids. EPA and DHA are well known to be beneficial for human health, and according to numerous recent clinical studies, may promote healthy heart, brain and visual function, and may also contribute to reducing inflammation and blood TGs. Krill is a natural source of phospholipids and OM3 fatty acids. The EPA and DHA contained in CaPre are delivered as a combination of OM3s as free fatty acids and OM3s bound to phospholipid esters. Both forms allow these PUFAs to reach the small intestine where they undergo rapid absorption and transformation into complex fat molecules that are required for lipid transport in the bloodstream. We believe that EPA and DHA are more efficiently transported by phospholipids sourced from krill oil than the EPA and DHA contained in fish oil that are transported either by TGs (as in dietary supplements) or as ethyl esters in other prescription OM3 drugs (such as LOVAZA and VASCEPA), which must then undergo additional digestion before they are ready for transport into the bloodstream. The digestion and absorption of OM3 ethyl ester drugs requires a particular enzymatic process that is highly dependent on the fat content of a meal – the higher the fat content, the better the OM3 ethyl ester absorption. High fat content meals are not recommended in patients with HTG. We believe that CaPre’s superior absorption profile could represent a significant clinical advantage, since taking it with a low-fat meal represents a healthier and more realistic regimen for patients with HTG who must follow a restricted low-fat diet.
CaPre is intended to be used as a therapy combined with positive lifestyle changes, such as a healthy diet and exercise, and can be administered either alone or with other drug treatment regimens such as statins (a class of drug used to reduce LDL-C). CaPre is intended to be taken orally once or twice per day in capsule form.
| - 25 - |
Potential Market for CaPre
We believe a significant opportunity exists for OM3 market expansion because, among other things:
| · | cardiovascular diseases, or CVD, and stroke are the leading causes of morbidity and mortality in the United States. The burden of CVD and stroke in terms of life-years lost, diminished quality of life, and direct and indirect medical costs also remains enormous; |
| · | evidence suggests potential for OM3s in other cardiometabolic indications; and |
| · | based on the assumption that the REDUCE-IT trial sponsored by Amarin and the STRENGTH trial sponsored by Astra Zeneca, or the CV outcome trials, will be positive, key opinion leaders interviewed by DP Analytics in the study described further below estimated that they would increase their own prescribing of OM3s by 42% in patients with high TGs (blood levels between 200 – 499 mg/dL) and by 35% in patients with severe HTG. |
According to the American Heart Association, the prevalence of HTG in the United States and globally correlates to the aging of the population and the increasing incidence of obesity and diabetes. Market participants, including the American Heart Association, have estimated that one-third of adults in the United States have elevated levels of TGs (TGs >150 mg/dL), including approximately 36 million people diagnosed with high HTG, and 3 to 4 million people diagnosed with severe HTG. Moreover, according to Ford, Archives of Internal Medicine in a study conducted between 1999 and 2004, 18% of adults in the United States, corresponding to approximately 40 million people, had elevated TG levels equal to or greater than 200 mg/dl, of which only 3.6% were treated specifically with TG-lowering medication. We believe this data indicates there is a large underserved market opportunity for CaPre.
In 2015, CaPre’s target market in the United States for severe HTG was estimated by IMS NSP Audit data to be approximately $750 million, with approximately 5 million prescriptions written annually over the prior four years. The total global market was estimated by GOED Proprietary Research in 2015 to be approximately $2.3 billion. We believe there is the potential to greatly expand the treatable market in the United States to the approximately 36 million people with high HTG, assuming favorable results from the CV outcome studies that are currently ongoing. These CV outcome trials are expected to report in mid-2018 (the REDUCE-IT trial sponsored by Amarin) and 2019 (the STRENGTH trial sponsored by Astra Zeneca) and are designed to evaluate the long-term benefit of lowering TGs on cardiovascular risks with prescription drugs containing OM3 fatty acids. If these trials are successful, additional clinical trials would likely be required for CaPre to also expand its label claims to the high HTG segment. Given the large portion of the adult population in the United States that have elevated levels of TGs but who go largely untreated, we believe there is the potential for a very significant increase in the total number of patients eligible for treatment if the CV outcome trials are positive.
CaPre has two FDA-approved and marketed branded competitors (LOVAZA and VASCEPA). In addition, Astra Zeneca has an FDA-approved product, EPANOVA, which has not yet been launched. LOVAZA generics became available on the U.S. market in 2013. In spite of generic options, audited prescription data from IMS NSP Audit data suggests that over 50% of OM3 prescriptions are written for branded products (LOVAZA or VASCEPA). By 2015, there had been only an approximately 25% decline in total market value, in spite of some generic switching that occurs at pharmacies. This stability of branded products is due in part to the fact that the pricing differential between branded and generic OM3 products is smaller than is typically the case between branded and generic products in the pharmaceutical industry. Based on both primary market research with pharmacy benefit managers, or PBMs, and audited prescription reports, the average pricing of generics is currently approximately $160 per month, while pricing for branded products averages $250 - $300 per month. Amarin has raised prices for VASCEPA annually since its launch in late 2013. PBMs offer “Preferred Brand” status (Tier 2 or Tier 3), without significant restrictions (i.e. no prior authorization, step edits, or high co-payments) for these branded OM3s.
Except as otherwise indicated, all of the information that follows under this heading has been derived from secondary sources, including audited U.S. prescribing data, and from a qualitative U.S. commercial and primary market research assessment conducted for us by DP Analytics, A Division of Destum Partners, Inc., or Destum, a market research firm, dated August 19, 2016, which we refer to as the Destum Market Research. In its market analysis for CaPre, Destum utilized secondary market data and reports and conducted primary qualitative market research with physicians and third-party payers, such as PBMs. One-on-one in-depth phone interviews lasting on average 30-60 minutes were conducted with 22 physicians and 5 PBMs, and key qualitative data was obtained by Destum on current clinical practice for treating patients with HTG, and their perceptions of the current unmet medical need in treating patients with HTG. All interviews were conducted by the same individual at Destum and recorded to ensure consistency and collection of key data points. Destum utilized OM3 prescription data from 2009 to 2015 to estimate the size of CaPre’s potential market. Based on its discussions with the PBMs, Destum also assumed CaPre would be viewed favorably by payers at launch (e.g., Tier 2 or 3, depending on payer plan, which is comparable to LOVAZA and VASCEPA). Upon completing the screening questionnaire and being approved for inclusion in Destum’s study, key opinion leaders, or KOLs, and high volume prescribers, or HVPs, were provided with a study questionnaire and were asked to comment on a target profile for a potential new OM3 “Product X” offering a “trifecta” of cardio-metabolic benefits similar to the potential efficacy and safety benefits demonstrated by CaPre in our two Phase 1 pharmacokinetic studies and two Phase 2 clinical trials, which we refer to as the Target Product Profile. Respondents were told that the unidentified product was being prepared for a TRILOGY Phase 3 program designed to confirm with statistical significance the product’s safety and efficacy in patients with severe HTG. The Target Product Profile was used by Destum strictly for market research analysis purposes and should not be construed as an indication of future performance of CaPre and should not be read as an expectation or guarantee of future performance or results of CaPre, and will not necessarily be an accurate indication of whether or not such results will be achieved by CaPre in our planned TRILOGY Phase 3 program. We subsequently retained Destum as our exclusive advisor and business development consultant to identify potential strategic partners for CaPre, under which Destum may be entitled to a success fee if a business arrangement or transaction is consummated. Destum’s market research and its conclusions were substantially completed prior our entry into this agreement with Destum.
| - 26 - |
During the Destum Market Research, KOLs and HVPs interviewed by Destum were asked to assess the level of unmet medical need associated with treating patients with severe HTG based on currently available treatment options. 91% of physicians interviewed by Destum indicated that they believe that the current unmet medical need for treating HTG was moderate to high. The reasons identified by these physicians for their dissatisfaction with the currently available OM3s included insufficient lowering of TGs (principally relating to VASCEPA), negative LDL-C effects (principally relating to LOVAZA), gastrointestinal side effects, and the fishy taste from fish oil-derived OM3s. Despite the availability of other drug classes to treat severe HTG, interviewed physicians indicated that they would welcome the introduction of new and improved OM3 products, particularly if they can address these perceived deficiencies.
Interviewed physicians responded favorably in the Destum Market Research to the Target Product Profile. They indicated that their weighted prescribing percentages of the Target Product Profile would increase by approximately 35% to 53% (with the range depending on the specific profile presented) in the severe HTG patient population within two years of the Target Product Profile’s approval. Approximately 60% of the interviewed physicians indicated that they would switch primarily due to the “trifecta effect” of the Target Product Profile on reducing TGs and LDL-C while elevating HDL-C, and the remaining 40% indicated they would switch primarily due to the Target Product Profile’s effective reduction of TGs alone. In connection with their responses, the interviewed physicians were instructed to assume the Target Product Profile and all currently available OM3 products were not subject to any reimbursement or coverage hurdles (e.g., all products were on an equal health care coverage playing field). This assumption was supported by our interviews with leading PBMs in the United States.
We plan to conduct additional market research with KOLs, HVPs, primary care physicians and payers to further develop and refine our understanding of the potential marketplace for CaPre.
Our Clinical Data
CaPre is being developed by us for the treatment of patients with severe HTG. In two Phase 2 clinical trials conducted by us in Canada (our COLT and TRIFECTA trials), CaPre was found to be safe and well-tolerated at all doses tested, with no serious adverse events that were considered treatment-related. Among the reported adverse events with an occurrence of greater than 2% of subjects and greater than placebo, only diarrhea had an incidence of 2.2%.
In both Phase 2 clinical trials, CaPre significantly lowered TGs in patients with mild to severe HTG. Importantly, in these studies, CaPre also demonstrated no deleterious effect on LDL-C (unlike LOVAZA and EPANOVA, which have been shown to significantly increase LDL-C in patients with severe HTG). Further, our Phase 2 data indicated that CaPre may actually reduce LDL-C. LDL-C is undesirable because it accumulates in the walls of blood vessels, where it can cause blockages (atherosclerosis). In the Phase 2 trials, CaPre also reduced non-HDL-C (all cholesterol contained in the bloodstream except HDL-C), which is also considered to be a marker of cardiovascular disease. The COLT trial data showed a mean increase of 7.7% in HDL-C with CaPre at 4 grams per day (p=0.07). Further studies in our planned TRILOGY Phase 3 program are required to demonstrate CaPre’s statistical significance with HDL-C.
We believe that these multiple potential cardiovascular benefits, if confirmed in our planned TRILOGY Phase 3 program, could be significant differentiators for CaPre in the marketplace, as no currently approved OM3 drug has shown an ability to positively modulate these four major blood lipid categories (TGs, non-HDL-C, LDL-C and HDL-C) in the treatment of severe HTG. We also believe that if supported by additional clinical trials, CaPre has the potential to become the best-in-class OM3 compound for the treatment of mild to moderate HTG.
On September 14, 2016, we announced positive data from our completed comparative bioavailability study, or the Bridging Study. The Bridging Study was an open-label, randomized, four-way, cross-over, bioavailability study comparing CaPre, given as a single dose of 4 grams in fasting and fed (high-fat) states, as compared to the FDA-approved HTG drug LOVAZA (OM3-acid ethyl esters) in 56 healthy volunteers. The protocol was reviewed and approved by the FDA. The primary objective of the Bridging Study was to compare the bioavailability of CaPre to LOVAZA, each administered as a single 4-gram dose with a high-fat meal, which is the condition under which administration of OM3 drugs will yield the highest levels of EPA and DHA in the blood, and therefore has the highest potential for toxicity. To allow us to rely on the long-term safety data of LOVAZA to support a 505(b)(2) NDA for CaPre, our results had to show that the blood levels of EPA and DHA resulting from a single 4-gram dose of CaPre are not significantly higher than from a single 4-gram dose of LOVAZA under fed (high-fat meal) conditions. The Bridging Study met all of its objectives and demonstrated that the levels of EPA and DHA following administration of CaPre did not exceed corresponding levels following administration of LOVAZA in subjects who were fed a high-fat meal. We expect that these results will support a claim by us that CaPre and LOVAZA have a comparable safety profile. Also, among subjects in a fasting state, CaPre demonstrated better bioavailability than LOVAZA, as measured by significantly higher blood levels of EPA and DHA. Since most HTG patients must follow a restricted low-fat diet, we believe that CaPre’s strong bioavailability profile could provide a more effective clinical solution for these patients.
| - 27 - |
We summarized and submitted data from our Bridging Study to the FDA for review and discussed it with the FDA at an End of Phase 2 meeting during the first quarter of 2017. We also presented our Bridging Study data at the National Lipid Association Conference in May 2017 and we plan to submit the data from our Bridging Study for peer review and publication.
The graph below illustrates that the Bridging Study achieved all of its objectives:
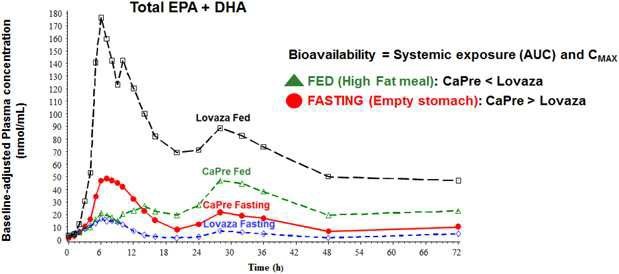
Absorption of EPA and DHA as ethyl ester formulations in the currently available prescription OM3 drugs derived from fish oil (such as LOVAZA and VASCEPA) require the breakdown of the ethyl esters by pancreatic enzymes (lipases) to be released into the blood. These particular enzymes are produced in response to the consumption of high-fat content meals, leading to optimal absorption of EPA and DHA. As a result, these OM3 ethyl ester formulations have demonstrated lower absorption and bioavailability when taken with a low-fat meal or on an empty stomach. As shown in our CAP13-101 study described further below, absorption of CaPre, which is formulated as OM3 phospholipids and free fatty acids, is not meaningfully affected by the fat content of a meal consumed prior to drug administration. Since a low-fat diet is typically a critical component for treatment of patients with severe HTG, we believe that being able to effectively combine CaPre with a low-fat diet could give CaPre a significant clinical and marketing advantage over the ethyl ester-based OM3s, such as LOVAZA and VASCEPA, that must be taken with a high-fat meal to achieve optimal absorption.
Our CAP13-101 study was an open-label, randomized, multiple-dose, single-center, parallel-design study in healthy volunteers. 42 subjects were enrolled into 3 groups of 14 subjects who took 1 gram, 2 grams or 4 grams of CaPre, administered once a day 30 minutes after breakfast. The objectives of the study were to determine the pharmacokinetic, or PK, profile and safety on Day 1 following a single oral dose and Day 14 following multiple oral doses of CaPre in individuals pursuing a low-fat diet (therapeutic lifestyle changes diet). The effect of a high-fat meal on the bioavailability of CaPre was also evaluated at Day 15. Blood samples were collected for assessment of EPA and DHA total lipids in plasma to derive the PK parameters.
The PK profile of CaPre following multiple 4-gram doses obtained in the CAP13-101 study at Day 14 was compared to the results obtained in a similar PK study (Offman 2013 - ECLIPSE 2) where LOVAZA was also administered at 4 grams a day for 14 days with a low-fat diet. Although CaPre contains approximately 2.5 times less EPA and DHA compared to LOVAZA (approximately 310 mg/1g capsule for CaPre versus 770 mg/1g capsule for LOVAZA), when administered with a low-fat meal, CaPre plasma levels of EPA and DHA are very similar to those of LOVAZA, as indicated by the area under the plasma drug concentration against time curve, or AUC, and the maximal plasma drug concentration. This study gives us confidence in the dosing and design of our planned TRILOGY Phase 3 program.
| - 28 - |
As illustrated by the two graphs below, CaPre reached similar blood and therapeutic levels to LOVAZA after 14 daily doses of CaPre at 4 grams/day, despite CaPre containing 2.5 times less EPA and DHA compared to LOVAZA:
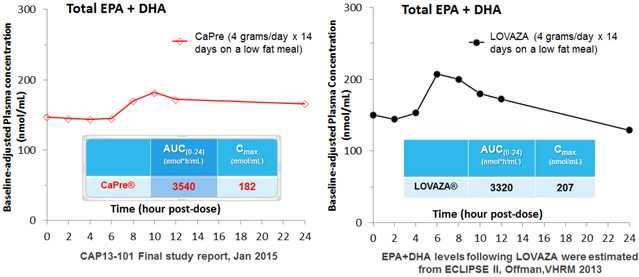
The graph below illustrates that the bioavailability of CaPre (total EPA+DHA levels in the blood) does not appear to be meaningfully affected by the fat content of a meal after multiple daily doses of CaPre at 4 grams/day (< 20% difference in AUC). We believe that CaPre’s strong bioavailability could represent a significant clinical advantage for CaPre since taking it with a low-fat meal represents a more realistic and attractive regimen for patients with HTG who must follow a restricted low-fat diet.
Our Study CAP13-101 CaPre Pharmacokinetics Shows No Significant Food Effect
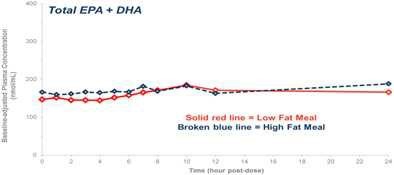
The graph below presents a summary of the effects of CaPre on patients’ lipid profiles as obtained in our completed TRIFECTA and COLT Phase 2 clinical trials. 90% of the patients in these clinical trials had mild to moderate HTG (levels between 200 - 499 mg/dL) and 10% of patients had severe HTG (levels between 500 and 877 mg/dL), which was the maximum level of TGs permitted by Health Canada’s study protocol. Only 30% of the participating patients were taking statins, which we believe is important because statins appear to enhance the TG-lowering effect of OM3s. In contrast, in our competitors’ summary data that follows, 100% of the patients in those studies with mild to moderate HTG were taking statins with their OM3s.
The summary data from our COLT and TRIFECTA clinical trials shows that CaPre significantly reduces TGs, but unlike some other prescription EPA/DHA-based OM3s, it has no deleterious effect on LDL-C and may potentially increase HDL-C (p=0.07), which we refer to as the “trifecta effect”. Also, a dose response was seen for all of the major lipid markers; the greater the dose of CaPre, the greater the beneficial effect of CaPre.
| - 29 - |
Our Phase 2 Study Results Show CaPre Dose Response and Potential for “Trifecta” Lipid Effect
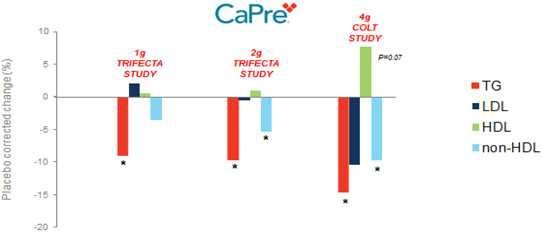
* Indicates results reached statistical significance
TRIFECTA for 1g (N=130) & 2g (N=128) and COLT for 4g (N=62). HDL-C results at 4g from COLT approached statistical significance at P=0.07.
We conducted a subgroup analysis including only patients with severe HTG, consisting of approximately 10% of the patients from our TRIFECTA study, to compare the effects of CaPre versus other OM3 drugs in the initial target population of patients with severe HTG. Despite being given at a lower dose (only 1 gram and 2 grams), CaPre’s results compared very well with data from independent studies for the other prescription OM3 drugs that are FDA-approved for the treatment of severe HTG at higher doses of 2 grams and 4 grams. While the results of this subgroup analysis were not statistically significant for CaPre (potentially due to the small sample size), numerically, the results compared well with the other OM3 drugs, even though CaPre was given at a much lower dose. The results for LDL-C, HDL-C and non-HDL-C levels in the subgroup shown in the table below are based on descriptive statistics only and are solely directional, meaning that no statistical testing was conducted and so no “p” values were generated.
Our Sub-Group Analysis in Patients with Severe HTG: CaPre1 at 1g and 2g Compares Well with Our Competitors2 at 2g and 4g
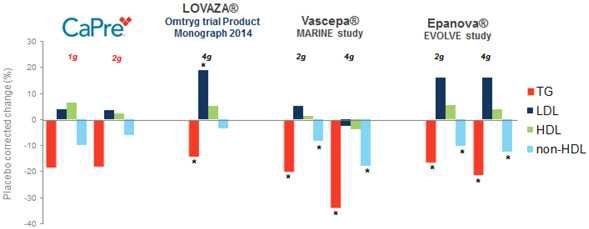
Only ~1/3 of all patients across all studies were on statins
* Indicates results reached statistical significance
1. Subgroup analysis on CaPre Phase 2 TRIFECTA study data in patients with severe HTG; (N=10 for 1g & N=14 for 2g). Results are not statistically significant for TGs, which may be explained by the small number of patients in this subgroup analysis. Results for LDL-C, HDL-C and non-HDL-C are based on descriptive statistics only (no statistical testing conducted).
2. LOVAZA 4g (N=103), VASCEPA 2g/4g (N=73/76), EPANOVA 2g/4g (N=100/99).
Since statins appear to enhance the TG-lowering property of OM3 drugs, we conducted a subgroup analysis that only included patients who were taking a statin at baseline in the COLT and TRIFECTA studies (approximately 30% of the population of both trials, combined). The graph below compares the TG-lowering effects of CaPre to other OM3s, all on a background of a statin drug, and shows that CaPre’s TG-lowering effects compare well with other FDA-approved OM3 drugs. We believe it is noteworthy that only 39 patients on 2 grams of CaPre in our TRIFECTA study (out of a total of 128) and only 22 patients on 4 grams of CaPre in our COLT study (out of 62) were taking statins.
| - 30 - |
The CaPre 2-gram bar graph in the table below shows the results from patients in our TRIFECTA trial who were taking statins. A statistically significant reduction in TGs (-25.7% placebo corrected) was seen in that statin subgroup. The CaPre 4-gram bar graph in the table below shows patient results only from our COLT trial (as there was no 4-gram component for our TRIFECTA). None of the results were statistically significant at 4 grams of CaPre, potentially due to the small number of patients (22) in the statins subgroup.
As seen in the larger full study analyses in the tables above, CaPre does not show any deleterious effect on LDL, and shows the potential to decrease LDL and increase HDL (p=0.07). These observations will need to be confirmed in our planned TRILOGY Phase 3 program.
Our Sub-Group Analysis in Patients Treated with Statins1 vs Independent Competitor Data2: Potential for CaPre Trifecta Effect
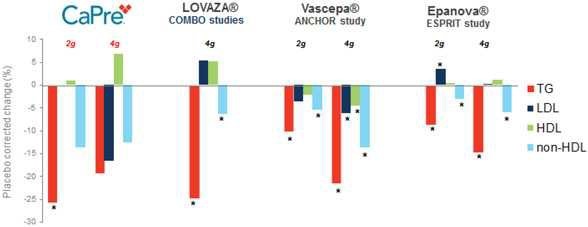
* Indicates results reached statistical significance
| 1. | CaPre subgroup analyses on patients treated with statins: TRIFECTA for 2g (N=39) and COLT for 4g (N=22). For CaPre 2g, results for LDL-C, HDL-C, and non-HDL-C are based on descriptive statistics only (no statistical testing was conducted). For CaPre 4g, no results are statistically significant which may be explained by the small number of patients. |
| 2. | All patients on a statin background: LOVAZA (N=122 for 4g), VASCEPA (N= 234 for 2g, N=227 for 4g), EPANOVA (N=209 for 2g, N=207 for 4g). Statins have been shown to enhance the efficacy of OM3 products – VASCEPA NDA 202057. Statistical review, section 4.2 ‘‘Other special/Subgroup populations’, p. 107; and Maki K et al. Clin. Ther. 2013 |
In summary, in addition to effectively reducing TG levels in patients with mild to severe HTG, clinical data collected by us to date indicates that CaPre may also have:
| • | beneficial effects on other blood lipids, such as HDL-C (good cholesterol) and non-HDL-C; |
| • | no deleterious effect on, and may potentially reduce, LDL-C (bad cholesterol) levels; and |
| • | absorption capability that is not meaningfully affected by the fat content of a meal consumed prior to drug administration, providing patients with the reassurance that following their physician-recommended low-fat diet will still result in high absorption. |
We believe that these features could set CaPre apart from currently available FDA-approved OM3 treatment options in the marketplace and could give us a significant clinical and marketing advantage.
| - 31 - |
CaPre’s potential clinical benefits as compared to currently available FDA-approved OM3 treatment options are summarized in the table below and indicate that CaPre may deliver a more complete lipid management solution for patients with severe HTG:
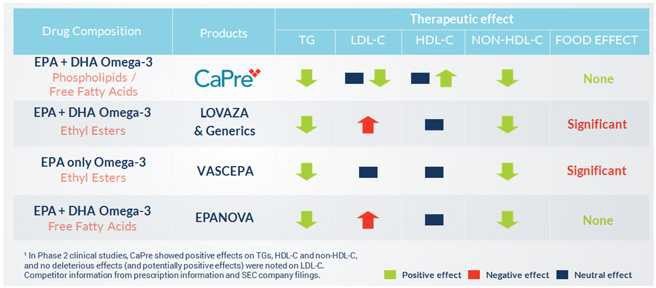
Our Nonclinical Research
In addition to our Phase 2 clinical trials, we carried out an extensive nonclinical program to demonstrate the safety of CaPre in a defined set of studies required by the FDA. These studies were carried out by contract research organizations with Good Laboratory Practice certification and conducted on various species of animals recommended by the FDA to investigate the long-term effects of CaPre at doses of up to 65 grams of human equivalent dose over 39 weeks. In these studies, hematological, biochemical, coagulation and overall health parameters of CaPre were evaluated and no toxic effects were observed in any of the segments of the studies. Other studies focused on the potential toxic effects of CaPre on vital systems, such as the cardiovascular, respiratory and central nervous system as evaluated by behavioral studies of the various species. These studies showed that CaPre did not have any adverse or toxic effects on any of the vital systems investigated, again up to doses well above the recommended clinical dose of CaPre. To rule out short term toxic effects of CaPre on genes, genomic toxicity studies were undertaken on accepted cellular and animal models. These studies showed no toxic effects of CaPre on any of the genetic markers indicative of potential gene altering toxic effects.
We believe the studies conducted to date indicate that CaPre is well-tolerated and shows no toxic effects on any of the physiological and vital systems of the tested animals or their genes or molecules at doses well above CaPre’s anticipated clinical therapeutic dose of 4 grams daily.
In parallel to our TRILOGY Phase 3 program, we will have to complete additional nonclinical studies, including a pre- and postnatal development study in rodents and a 26-week oral carcinogenicity study in transgenic homozygous rasH2 mice. These nonclinical studies will be required to support a NDA for CaPre.
Our TRILOGY Phase 3 Program Design
In March 2017, we announced our plans to proceed with our TRILOGY Phase 3 program following our End-of-Phase 2 meeting with the FDA in February 2017. Based on the guidance we have received from the FDA, we are now conducting two pivotal, randomized, placebo-controlled, double-blinded Phase 3 studies to evaluate the safety and efficacy of CaPre in patients with severe HTG. These studies of 26-week duration will evaluate CaPre’s ability to lower TGs from baseline in approximately 500 patients (approximately 250 per study) randomized to either 4 grams daily or placebo. The FDA’s feedback supported our plan to conduct two studies in parallel, potentially reducing the cost and shortening the time to an NDA submission. These studies will be conducted in approximately 150 sites across North America.
The primary endpoint of these studies is to determine the efficacy of CaPre at 4 grams/day compared to placebo in lowering TGs after 12 weeks in severe HTG patients, and to confirm safety. The study was designed to provide at least 90% statistical power to detect a difference of at least a 20% decrease from baseline in TGs between CaPre and placebo. In addition, the Phase 3 studies will include numerous secondary and exploratory endpoints, which are designed to assess the effect of CaPre on the broader lipid profile and certain metabolic, inflammatory and CV risk markers.
Late in 2017, based on feedback from the FDA, Acasti finalized its Chemistry, Manufacturing, and Controls plans and the clinical trial design that supports Acasti’s TRILOGY Phase 3 program. In parallel with the Phase 3 clinical trial planning, additional cGMP production lots of API (known as NKPL66) and CaPre were manufactured during the fourth quarter, enabling Acasti to continue to accumulate the CaPre and placebo inventory required to support the activation of clinical trial sites and patient randomization. Acasti also purchased additional raw krill oil material from Neptune to adequately supply the entire Phase 3 clinical program and to ensure sufficient material to prepare for validation and future commercial activities.
| - 32 - |
During the quarter ended December 31, 2017, we further advanced our clinical development of CaPre. We initiated TRILOGY, our Phase 3 clinical program and began site activation and patient enrollment at the end of 2017. We are working with a major clinical research organization to manage our TRILOGY Phase 3 program. Continued site activation, patient recruitment and enrollment, patient screening and randomization are now underway.
In November 2017, we announced that Dariush Mozaffarian, M.D., Dr.P.H., agreed to serve as the principal investigator of our Phase 3 clinical program. Dr. Mozaffarian is a cardiologist and epidemiologist serving as the Jean Mayer Professor of Nutrition & Medicine, and the Dean of the Friedman School of Nutrition Science & Policy at Tuft’s University. His widely published research focuses on how diets, such as those rich in OM3s and lifestyle influence cardiometabolic health, and how effective policies can improve health and wellness.
The following chart illustrates the expected design and dosing of our TRILOGY Phase 3 program for CaPre.
Planned Phase 3 Clinical Program
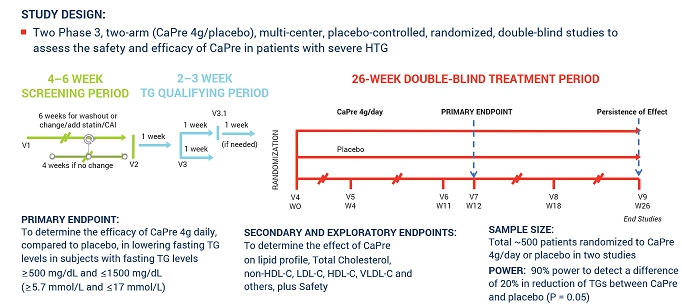
Clinical Trial Process and Timeline
During the second half of 2017, our clinical research organization, or CRO, began the process of identifying a sufficient number of clinical sites with experienced investigators to conduct the two Phase 3 clinical trials. Site activation involves negotiating a contract, gaining approval from the site’s Institutional Review Board, or IRB, and delivery of clinical supplies. It was determined that approximately 150 sites across North America will be used to randomize the total of nearly 500 patients with severe HTG required to complete the two Phase 3 studies. Site activation was initiated in the fourth quarter of 2017, and is currently ongoing. Site activation runs concurrently with patient screening and enrollment in order to secure an adequate number of sites to achieve the patient enrollment goals of the program.
Initiating a clinical trial involves numerous steps to engage investigators to screen and qualify patients as participants, prior to randomizing them to test the investigational drug. This entire screening and randomization process takes an average of six to nine weeks. Patient recruitment is conducted by each clinical trial site, supported by resources provided by the CRO. After a patient is identified by the investigator as a possible candidate for the clinical trial, they are screened to determine their eligibility for trial enrollment. The screening period takes four to six weeks. Patients must meet the inclusion criteria of the study, as described in the trial plan, also known as a protocol. We expect each patient will require two screening visits with the investigator’s clinical staff, whereby medical history and patient consent are obtained. This further qualification process takes two to three weeks.
When patient qualification is confirmed, the process of randomization begins. Approximately 245 patients should be randomized in each Phase 3 study. This sample size per study would provide 90% statistical power to detect at least a 20% decrease in TG levels from baseline to week 12 between CaPre and placebo with a two-sided α at 0.05 (primary endpoint), a difference that is believed to be clinically relevant. A randomized controlled trial is designed to reduce bias when testing an investigational treatment. The process of assigning patients to these groups by chance, rather than choice, is called randomization. The groups are referred to as the experimental group or the control group. In the Phase 3 clinical trials, patients will be assigned to either receive CaPre (experimental) or placebo (control). Each patient will be on CaPre or placebo for a period of 26 weeks.
| - 33 - |
The two Phase 3 clinical trials will proceed to dosing both the experimental and control groups, according to the protocol, to assess CaPre’s efficacy and safety compared to placebo. In these double-blind studies, neither the patients nor the investigator knows which treatment (experimental drug or placebo) a patient receives. Only after all data has been recorded and analyzed will such investigators and participants learn which were which. The trial conduct and patient safety are rigorously monitored to ensure regulatory compliance and to maintain the integrity of the study in order to assess outcomes.
We began patient randomization in the two Phase 3 trials in the first calendar quarter of 2018, and the two Phase 3 trials are expected to take approximately 18 months to complete. More specifically, the enrollment period takes approximately one year and the treatment period takes approximately 26 weeks per patient randomized. We plan to complete the program in mid-2019, and to report topline results from the parallel trials by the end of 2019.
Our Regulatory Strategy for CaPre
Our strategy is to develop and initially commercialize CaPre for the treatment of severe HTG. The TRILOGY Phase 3 program was initiated during the second half of 2017 and has been designed to evaluate the clinical effect of CaPre on TGs, non-HDL-C, LDL-C, and HDL-C levels together with a variety of other cardiometabolic biomarkers in patients with severe HTG.
In December 2015, we announced that we intend to pursue a 505(b)(2) regulatory pathway towards an NDA approval in the United States. A 505(b)(2) regulatory pathway is defined in the U.S. Federal Food Drug and Cosmetic Act (FDCA) as an NDA containing investigations of safety and effectiveness that are being relied upon for approval and were not, in whole, conducted by or for the applicant, and for which the applicant has not obtained a right of reference. 505(b)(2) regulatory pathways differ from a typical NDA because they allow a sponsor to rely, at least in part, on the FDA’s findings of safety and/or effectiveness for a previously- approved drug. We intend to pursue the 505(b)(2) regulatory pathway as a strategy to leverage the large body of safety data for LOVAZA, which could accelerate and streamline the development of CaPre and reduce associated costs and risks.
In connection with our intended use of the 505(b)(2) pathway, the FDA supported our proposal to conduct our Bridging Study that compared CaPre (which has an OM3 free fatty acid/phospholipid composition) with the FDA-approved OM3 drug LOVAZA (which has an OM3-acid ethyl esters composition) in healthy volunteers. In February 2017, we met with the FDA to review our Bridging Study data. We confirmed with the FDA the 505(b)(2) regulatory approach, which allows us to use the safety data for LOVAZA, and we finalized the study design for the two Phase 3 TRILOGY clinical trials, which will be required for NDA approval. The first clinical sites for our TRILOGY Phase 3 program (as described above), were initiated on schedule at the end of 2017, and the TRILOGY 1 and 2 trials are currently proceeding according to plan.
Our planned key milestones and development timeline are presented below.
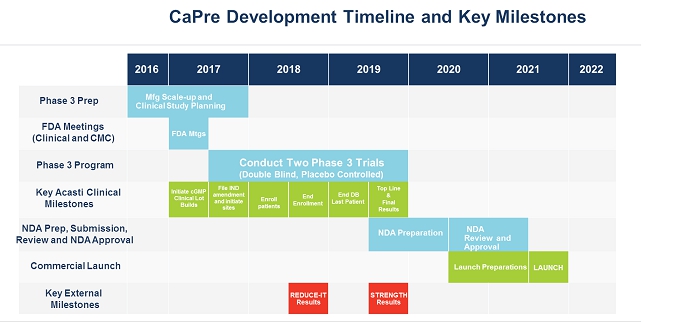
| - 34 - |
Our Intellectual Property Strategy
Under a license agreement we entered into with Neptune in August 2008, which was later amended on February 9, 2009 and March 7, 2013 (the “License Agreement”), we received an exclusive license to use certain intellectual property of Neptune (which includes several patents) to develop and commercialize CaPre and our novel and active pharmaceutical ingredients, or APIs, for use in pharmaceutical and medical food applications in the cardiometabolic field. The term of the License Agreement expires on the date of the last-to-expire licensed patents in 2022. As a result of a royalty prepayment transaction we entered into with Neptune on December 4, 2012, we are no longer required to pay any royalties to Neptune under the License Agreement during its term for the use of the licensed intellectual property.
On August 8, 2017, Neptune announced that it sold its krill oil inventory and intellectual property to Aker BioMarine Antarctic AS, or Aker. The sold intellectual property included the intellectual property to which rights were granted to Acasti under the License Agreement. As part of that transaction, Aker entered into a patent license agreement, or Aker Patent License Agreement, with Neptune pursuant to which it granted to Neptune the right to sublicense back to Acasti certain intellectual property as necessary to allow the Corporation to maintain its license grant under the original License Agreement. Accordingly, the license granted to the Corporation under the License Agreement remains in force.
Upon the expiry of our license agreement with Neptune, we believe that CaPre will be covered under our own issued and pending patents, and we do not believe that we will afterwards require any license from Neptune to support the commercialization of CaPre.
We continue to expand our own intellectual property, or IP, patent portfolio. We have filed patent applications in 23 jurisdictions, including with the European Patent Office (but excluding the individual countries where we have subsequently registered), and in countries in North America, Asia and Australia for our “Concentrated Therapeutic Phospholipid Composition”, or Proprietary Composition, to treat HTG. We currently have 22 issued or allowed patents and 18 patent applications pending.
Two U.S. patents, U.S. Patent Nos. 8,586,567 and 9,475,830, have issued which relate to the use of concentrated therapeutic phospholipid compositions for treating or preventing diseases associated with cardiovascular disease, comprising administering an effective amount of a concentrated therapeutic phospholipid composition. More specifically, U.S. Patent No. 8,586,567 covers a method of reducing serum TG levels comprising administering to a subject an effective amount of a concentrated phospholipid (PL) composition having, among other things, a concentration of total phospholipids in the composition of about 66% (w/w). U.S. Patent No. 9,475,830 covers a method of treating HTG comprising administering to a subject a therapeutically effective amount of a concentrated therapeutic phospholipid composition, having, among other things, a concentration of total phospholipids in the composition of about 60% (w/w). We also filed a U.S. continuation patent application (U.S. Patent Application Serial No. 15/258,044) to pursue claims directed towards a composition encompassing an extract comprising a PL content between about 60% to about 99%.
In 2017, additional patents were granted to us by the Taiwanese, Korean, and Australian patent offices to protect our Proprietary Composition using compositions of matter claims and medical use claims. In 2018, Acasti was also granted patents by the Canadian Intellectual Property Office, the European Patent Office (EPO), the Russian Patent Office, and the Japanese Patent Office for the Proprietary Composition all of which contain compositions of matter claims and medical use claims. Accordingly, patent protection for the Proprietary Composition has now been secured in for example Australia, Canada, China, Europe (including Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, United Kingdom, Italy, Netherlands, Norway, Portugal and Sweden), Japan, Korea, Russia, Saudi Arabia, Taiwan, the U.S. and South Africa.
A patent is generally valid for 20 years from the date of first filing. However, patent terms can be subject to extensions in some jurisdictions in order to compensate, for example, for delays caused by the patent office during prosecution of the patent application or for regulatory delays during the pre-market approval process.
We believe these patents and patent applications increase potential commercial opportunities for CaPre, including through possible licensing and partnership opportunities. We are committed to building a global portfolio of patents to ensure long-lasting and comprehensive intellectual property protection and to safeguard potentially valuable market expansion opportunities.
Our Australian patent No. 2010312238 was opposed by Enzymotec Ltd., but that opposition has been since been discontinued. Our patent No. 600167 in New Zealand, which is in force until 2030 and relates to a concentrated phospholipid composition comprising 60% PL and method of using the same for treating cardiovascular diseases, has been opposed by BIO-MER Ltd. The evidentiary stage in the New Zealand patent opposition has been completed. The next step is the Hearing. In our view, no new prior art has been presented that was not already considered in other jurisdictions, such as in the United States and Japan, where our patents are in force.
The trademark CaPre® is registered in the United States, Canada, Australia, China, Japan and Europe. In addition, we also protect our optimization and extraction processes through provisional patents, industrial trade secrets and know-how.
| - 35 - |
Manufacturing of CaPre
We are developing CaPre as a new chemical entity (which means a novel chemical product protected by patents), and we plan to conduct our TRILOGY Phase 3 program using good manufacturing practices, or cGMP, good clinical practices, or cGCP, and good laboratory practices, or cGLP.
The contract manufacturing organizations, or CMOs, selected by us for manufacturing and packaging are all cGMP compliant. In preparation for our TRILOGY Phase 3 program, working together with our pharmaceutical CMOs, we advanced the installation and qualification of the proprietary extraction and purification equipment used to manufacture CaPre. We ran our first scaled cGMP production lots of CaPre at CordenPharma’s Chenôve facility in Dijon, France during the first half of 2017. Batch sizes of 10 to 12 kilograms of CaPre have been successfully produced and tested clinically, and we scaled up to 100 kg/day in late 2017 to fulfill the clinical product requirements for our TRILOGY Phase 3 program and initial commercial launch. As of the date of this annual report, we have completed 9 clinical lots of NKPL66 and CaPre for our Phase 3 studies.
Our Business and Commercialization Strategy
Key elements of our business and commercialization strategy include initially obtaining regulatory approval for CaPre in the United States for severe HTG. We plan to launch CaPre ourselves in the US market. Our preferred strategy outside the United States is to commercialize CaPre through regional or country-specific strategic partnerships, and to potentially seek support and funding from each partner for in-country clinical development, registration and commercialization activities. We believe that a late development-stage and differentiated drug candidate like CaPre could be attractive to various global, regional or specialty pharmaceutical companies, and we are taking a targeted approach to partnering and licensing in various geographies. We also recently hired a Chief Commercial Officer who is chartered with developing and implementing our ex-US partnering strategies, as well as the US launch planning and execution. See “Recent Developments”.
Our key commercialization goals include:
| · | complete our TRILOGY Phase 3 program and, assuming the results are positive, file a new drug application, or NDA, to obtain regulatory approval for CaPre in the United States, initially for the treatment of severe HTG, with the potential to afterwards expand CaPre’s indication to the treatment of high TGs (although at least one additional clinical trial would likely be required to expand CaPre’s indication to this segment); |
| · | continue to strengthen our patent portfolio and other intellectual property rights; |
| · | continue planning for the potential launch of CaPre in the United States; and |
| · | continue to pursue strategic opportunities outside of the United States, such as licensing or similar transactions, joint ventures, partnerships, strategic alliances or alternative financing transactions, to provide development capital, market access and other strategic sources of capital. |
In addition to completing our TRILOGY Phase 3 program, we expect that additional time and capital will be required to complete the filing of an NDA to obtain FDA approval for CaPre in the United States, and to complete business development collaborations, marketing and other pre-commercialization activities before reaching the commercial launch of CaPre.
Competition
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to CaPre. We believe that the number of companies seeking to develop products and therapies similar to CaPre will likely increase, particularly if the CV outcome trials by Amarin and/or Astra Zeneca are successful.
Our competitors in the United States and globally include large, well-established pharmaceutical companies, specialty pharmaceutical sales and marketing companies, and specialized cardiovascular treatment companies. GlaxoSmithKline plc, which currently sells LOVAZA, a prescription-only OM3 fatty acid indicated for patients with severe HTG, was approved by the FDA in 2004 and has been available in the U.S. market since 2005. Multiple generic versions of LOVAZA are now available in the United States. Amarin launched its prescription-only OM3 drug VASCEPA in 2013, and reached a market share of approximately 20% by the end of 2015. In addition, EPANOVA (OM3-carboxylic acids) capsules, a free fatty acid form of OM3 (comprised of 55% EPA and 20% DHA), is FDA-approved for patients with severe HTG. Omtryg, another OM3-acid fatty acid composition developed by Trygg Pharma AS, received FDA approval for severe HTG. Neither EPANOVA nor Omtryg have yet been commercially launched, but could launch at any time. Other large companies with products that would compete indirectly with CaPre include AbbVie, Inc., which currently sells Tricor and Trilipix for the treatment of severe HTG, and Niaspan, which is primarily used to raise HDL-C but is also used to lower TGs. Generic versions of Tricor, Trilipix, and Niaspan are also now available in the United States. In addition, we are aware of a number of other pharmaceutical companies that are developing products that, if approved and marketed, would compete with CaPre.
| - 36 - |
Raw Materials
We use semi-refined raw krill oil as our primary raw material to produce CaPre. Krill is generally harvested in Antarctic waters. The total quantity of the krill species is estimated to be at least 500,000,000 metric tons. The krill biomass is the world’s most abundant biomass and is monitored to help ensure sustainable cultivation. Historically, we have sourced all of our krill oil from Neptune. On August 8, 2017, Neptune announced its near-term plan to discontinue krill oil production and the sale of its krill oil inventory and intellectual property to Aker. In the three-month period ending December 31, 2017, we purchased a reserve of krill oil from Neptune that will be used in the production of CaPre capsules for our Phase 3 clinical trials. We believe that alternative supplies of krill oil that can meet our specifications will be readily available and we are currently evaluating alternative suppliers of krill oil. At March 31, 2018, a reserve of krill oil was stored at Neptune’s facility located in Sherbrooke, Québec.
Employees, Specialized Skills and Knowledge
Our management consists of professionals from business development, sales and marketing, clinical development, pharmaceutical manufacturing, finance and science backgrounds. Our research team includes scientists with expertise in pharmaceutical development, chemistry, manufacturing and controls, nonclinical and clinical studies, pharmacology, regulatory affairs, quality assurance/quality control, intellectual property and strategic alliances. We currently employ 24 full-time employees with the majority working out of the Corporation’s headquarters in Laval and its laboratory in Sherbrooke. We generally require all of our employees to enter into invention assignment, non-disclosure and non-compete agreements. We rely, in part, on some administrative and general accounting support from Neptune, and we also rely on third-party consultants from time to time. Our employees are not covered by any collective bargaining agreement or represented by a trade union.
Additional Information About Our Phase 2 Clinical Trials
Our COLT Trial
Our COLT clinical trial, which was completed in 2014, was a randomized, open-label, dose-ranging, multi-center trial in Canada designed to assess the safety and efficacy of CaPre in the treatment of patients with TG levels between 200-877 mg/dL. The primary objectives of the COLT study were to evaluate the safety and efficacy of 0.5 gram, 1 gram, 2 grams and 4 grams of CaPre per day in reducing fasting plasma TGs over 4 and 8 weeks, as compared to the standard of care alone.
The secondary objectives of the COLT study were to evaluate:
| · | the effect of CaPre on fasting plasma TGs in patients with TGs between 200-499 mg/dL (mild to moderate HTG); |
| · | the dose dependent effect on fasting plasma triglycerides in patients with TGs between 500-877 mg/dL (severe HTG); and |
| · | the effect of CaPre on fasting plasma levels of LDL-C (direct measurement), HDL-C, non-HDL-C, hs-CRP and OM3 index. |
The final results of the COLT trial indicated that CaPre was safe and effective in reducing TGs in patients with mild to severe HTG with significant mean (average) TG reductions above 20% after 8 weeks of treatment with daily doses of 4 grams and 2 grams. Demographics and baseline characteristics of the patient population were balanced in terms of age, race and gender. A total of 288 patients were enrolled and randomized and 270 patients completed the study, which exceeded our targeted number of evaluable patients. From this patient population, approximately 90% had mild to moderate HTG.
The proportion of patients treated with CaPre that experienced one or more adverse events in the COLT trial was similar to that of the standard of care group (30.0% versus 34.5%, respectively). A substantial majority of adverse events were mild (82.3%) and no severe treatment-related adverse effects were reported. Only one patient was discontinued from the study due to an adverse event of moderate intensity. While the rate of gastrointestinal side effects was higher in the CaPre groups compared to standard of care alone and appeared to increase in a dose-related manner, none of the subjects participating in the study suffered from a serious adverse event. The COLT study results showed that even at higher doses, CaPre is safe and well tolerated with only transient and predominantly mild adverse events occurring at low rates.
The COLT trial met its primary objective of showing CaPre to be safe and effective in reducing TGs in patients with mild to severe HTG. After only a 4-week treatment, CaPre achieved a statistically significant TG reduction as compared to standard of care alone. Standard of care could be any treatment physicians considered appropriate in a real-life clinical setting and included lifestyle modifications as well as statins and/or ezetimibe. Patients treated with 4 grams of CaPre per day over 4 weeks reached a mean TG decrease of 15.4% from baseline and a mean improvement of 18.0% over the standard of care. Results also showed increased benefits after 8 weeks of treatment, with patients on a daily dose of 4 grams of CaPre registering a mean TG decrease of 21.6% from baseline and a mean improvement of 14.4% over the standard of care.
| - 37 - |
After 8 weeks of treatment, patients treated with 1 gram of CaPre for the first 4 weeks of treatment and 2 grams for the following 4 weeks, showed a statistically significant TG mean improvement of 16.2% over the standard of care, corresponding to a 23.3% reduction for the 1-2 grams patient population as compared to a 7.1% reduction for the standard of care. After 8 weeks of treatment, patients treated with 2 grams of CaPre for the entire 8 weeks showed statistically significant TG mean improvements of 14.8% over the standard of care, corresponding to a 22.0% reduction for the 2 grams as compared to a 7.1% reduction for the standard of care. Also, after 8 weeks of treatment, patients treated with 4 grams for the entire 8 weeks showed statistically significant TG, non-HDL-C and HbA1C mean improvements of 14.4% and 9.8% and 15.0%, respectively, as compared to standard of care. The 4-gram group showed mean improvements in:
| · | TGs of 14.4%, corresponding to a reduction of 21.6% as compared to a reduction of a 7.1% for the standard of care group, |
| · | non-HDL-C of 9.8%, corresponding to a reduction of 12.0% as compared to a reduction of 2.3% for the standard of care group, and |
| · | HbA1C of 15.0%, corresponding to a reduction of 3.5% as compared to an increase of 11.5% for the standard of care group. |
In addition, all combined doses of CaPre showed a statistically significant treatment effect on HDL-C levels, with an increase of 7.4% as compared to standard of care. Trends (p-value < 0.1) were also noted on patients treated with 4 grams of CaPre for the entire 8-week treatment period with mean reduction of total cholesterol of 7.0% and increase of HDL-C levels of 7.7%, as compared to the standard of care. The results of the COLT trial indicated that CaPre has no significant deleterious effect on LDL-C (bad cholesterol) levels.
Our TRIFECTA Trial
Our TRIFECTA clinical trial, which was completed in 2015, was a 12-week, randomized, placebo-controlled, double-blind, dose-ranging trial in Canada, designed to assess the safety and efficacy of CaPre at a dose of 1 gram or 2 grams on fasting plasma TGs as compared to a placebo in patients with TG levels between 200-877 mg/dL. A total of 387 patients were randomized and 365 patients completed the 12-week study, consistent with our targeted number of evaluable patients. From this patient population, approximately 90% had mild to moderate HTG with baseline TGs between 200 and 499 mg/dL. The remainder had severe HTG with baseline TGs between 500 and 877 mg/dL. Approximately 30% of patients were on lipid-lowering medications, such as statins, and approximately 10% were diabetic.
Similar to our COLT study, the primary objective of the TRIFECTA study was to evaluate the effect of CaPre on fasting plasma TGs in patients with TGs between 200-877 mg/dL and to assess the tolerability and safety of CaPre. The secondary objectives of the TRIFECTA study were to evaluate:
| · | the effect of CaPre on fasting plasma TGs in patients with TGs between 200-499 mg/dL; |
| · | the dose dependent effect on fasting plasma TGs in patients with TGs between 500-877 mg/dL; and |
| · | the effect of CaPre in patients with mild to moderate HTG and severe HTG on fasting plasma levels of LDL-C (direct measurement), and on fasting plasma levels of HDL-C, non-HDL-C, hs-CRP and OM3 index. |
CaPre successfully met the TRIFECTA’s study’s primary objective. The placebo-corrected percentage change in TGs were decreases of 9.1% (p=0.049) and 9.7% (p=0.044) for 1 gram and 2 grams of CaPre, respectively. Key secondary objectives were also met:
| · | there was a statistically significant decrease in non-HDL-C versus placebo (p=0.038), with the 2-gram group decreasing by 5.3% from baseline versus placebo over the 12-week period; |
| · | HDL-C (good cholesterol) slightly increased at both the 1-gram and 2-gram levels; and |
| · | LDL-C (bad cholesterol) and slightly decreased at the 2-gram level. |
Finally, a statistically significant dose response increase in the OM3 index for patients on 1 gram and 2 grams versus placebo was noted. The OM3 index reflects the percentage of EPA and DHA in red blood cell fatty acids and the risk of cardiovascular disease is considered to be lower as the OM3 index increases.
CaPre was found to be safe and well tolerated at all doses tested, with no serious adverse events that were considered treatment- related. Out of 387 randomized patients, a total of 7 (1.8%) were discontinued as a result of adverse events, three were on placebo, two were on 1 gram and two were on 2 grams. The predominant incidence was gastrointestinal-related, with no difference between CaPre and placebo. The safety profiles of patients on CaPre and placebo were similar.
The COLT and TRIFECTA clinical trials were conducted by JSS Medical Research, a CRO specializing in the pharmaceutical, biotechnology, nutraceutical and medical device industries, which is both owned and managed by Dr. John Sampalis, the brother of Dr. Tina Sampalis, who previously was our President and Chief Global Strategy Officer. JSS was selected by us following a rigorous due diligence process. Our board of directors appointed an external independent auditor, SNC Lavalin Pharma, to confirm and validate the clinical trials’ achievements, milestones and payments.
| - 38 - |
Government Regulation
United States Drug Development
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record- keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug products such as CaPre. Generally, before a new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority.
FDA Regulatory Process
In the United States, the FDA regulates drugs under the FDCA and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state and local statutes and regulations require the expenditure of substantial time and financial resources.
In order to be marketed in the United States, CaPre must be approved by the FDA through the NDA review process. The process required before a drug may be marketed in the United States generally involves the following:
| · | completion of extensive nonclinical (animal) and formulation studies in accordance with applicable regulations, including the FDA’s Good Laboratory Practice, or GLP, regulations; |
| · | submission of an investigational new drug, or IND, which must become effective before human clinical trials may begin in the United States; |
| · | performance of adequate and well-controlled clinical trials in accordance with the applicable IND and other clinical study- related regulations, such as current Good Clinical Practices, to establish the safety and efficacy of the proposed drug for its proposed indication; |
| · | submission of an NDA for a new drug; |
| · | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| · | satisfactory completion of potential FDA audit of the nonclinical and/or clinical trial sites that generated the data in support of the NDA; and |
| · | FDA review and approval of the NDA prior to any commercial marketing or sale of the drug in the United States. |
The data required to support an NDA is generated in two distinct development stages: nonclinical and clinical. The nonclinical development stage generally involves synthesizing or otherwise producing the active component, developing the formulation and determining the manufacturing process, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies in the laboratory, which support subsequent clinical testing. The sponsor must submit the results of the nonclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND, which is a request for authorization from the FDA to administer an investigational drug product to humans. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials. The FDA may also place the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. A clinical hold may be imposed at any time before or during a clinical trial due to safety concerns or non-compliance.
The clinical stage of development first involves the administration of the investigational drug to healthy volunteers and then to patients with the disease being targeted with the drug, all done under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with cGCP. All research subjects must provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, data collection, and the parameters to be used to monitor subject safety and assess the investigational drug’s efficacy. Each protocol, and any subsequent amendments to the protocol or new investigator’s information, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or its legal representative. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries, as well as reporting of safety information under the IND.
| - 39 - |
Clinical studies are generally conducted in three sequential phases that may overlap, known as Phase 1, Phase 2 and Phase 3 clinical trials. Phase 1 generally involves a small number of healthy volunteers who are initially exposed to a single dose and then multiple doses of the investigational drug. The primary purpose of these studies is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. Phase 2 trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks and preliminary evaluation of efficacy. Phase 3 clinical trials generally involve large numbers of patients at multiple sites, often in multiple countries (from several hundred to several thousand subjects) and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use, and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. Phase 3 clinical trials should, if possible, include comparisons with placebo and may include a comparison to approved therapies. The duration of treatment is often extended to mimic the actual use of a product during marketing. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA (Pivotal Studies).
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA. In addition, written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk.
Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides oversight and will determine whether or not a trial may move forward at designated check points based on review of interim data from the study. A clinical trial may be terminated or suspended based on evolving business objectives and/or competitive climate.
The manufacturing process must be capable of consistently producing quality batches of the investigational drug and, among other things, must develop methods for testing the identity, strength, quality and purity of the final drug product. The sponsor must develop appropriate labeling that sets forth the conditions of intended use. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Post-approval studies, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 studies as part of a post-approval commitment, such as pediatric studies.
NDA and FDA Review Process
Nonclinical and clinical information is filed with the FDA in an NDA along with proposed labeling. The NDA is a request for approval to market the drug and must contain proof of safety, purity, potency and efficacy, which is demonstrated by extensive nonclinical and clinical testing. Data may come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product to the satisfaction of the FDA.
The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances. FDA approval of an NDA must be obtained before marketing a drug in the United States. In addition, under the Pediatric Research Equity Act, an NDA or supplement to an NDA must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has ten months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant. This review typically takes 12 months from the date the NDA is submitted to the FDA including the screening which takes a period of 60 days. The FDA does not always meet its PDUFA goal dates for standard NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions with the FDA.
| - 40 - |
Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with cGCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities, it will issue a Complete Response Letter, or CRL. A CRL indicates that the review cycle of the application is complete and whether the application is approved and, when applicable, the CRL describes the specific deficiencies in the NDA and may require additional clinical data and/or an additional Phase 3 clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. The applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval.
If a product receives marketing approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling, may condition the approval of the NDA on other changes to the proposed labeling, or may require a Risk Evaluation and Mitigation Strategy (REMS), which could limit the ability to market the drug once approved. The FDA may also require the development of adequate controls and specifications, or a commitment to conduct post-market testing or clinical trials and surveillance to monitor the effects of approved products.
U.S. Post-Marketing Requirements
Following approval of a new product, a pharmaceutical company and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling, or “off-label use”, limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers and distributors may not market or promote such off-label uses. Modifications or enhancements to the product or its labeling or changes of the site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process. In some cases, these changes will require the submission of clinical data and the payment of a user fee.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of our prescription drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing and review of the relevant NDA.
Non-U.S. Drug Regulation
In Canada, biopharmaceutical product candidates are regulated by the Food and Drugs Act and the related rules and regulations, which are enforced by the Therapeutic Products Directorate of Health Canada. In order to obtain approval for commercializing new drugs in Canada, the sponsor must satisfy many regulatory conditions. The sponsor must first complete preclinical studies in order to file a clinical trial application, or CTA, in Canada. The sponsor will then receive different clearance authorizations to proceed with Phase I clinical trials, which can then lead to Phase 2 and Phase 3 clinical trials. Once all three phases of trials are completed, the sponsor must file a registration file named a New Drug Submission, or NDS, in Canada. If the NDS demonstrates that the product was developed in accordance with the regulatory authorities’ rules, regulations and guidelines and demonstrates favorable safety and efficacy and receives a favorable risk/benefit analysis, then the regulatory authorities issue a notice of compliance, which allows the sponsor to market the product.
| - 41 - |
In addition to regulations in the United States and Canada, we are subject to a variety of regulations governing clinical studies and commercial sales and distribution of our products in other jurisdictions around the world. These laws and regulations typically require the licensing of manufacturing and contract research facilities, carefully controlled research and testing of product candidates and governmental review and approval of results prior to marketing therapeutic product candidates. Additionally, they require adherence to good laboratory practices, good clinical practices and good manufacturing practices during production. The process of new drug approvals by regulators in the United States, Canada and the European Union are generally considered to be among the most rigorous in the world.
Whether or not the FDA or Health Canada approval is obtained for a product, we must obtain approval from the comparable regulatory authorities of other countries before we can commence clinical studies or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for the FDA or Health Canada approval. The requirements governing the conduct of clinical studies, product licensing, pricing and reimbursement vary greatly from country to country. In some international markets, additional clinical trials may be required prior to the filing or approval of marketing applications within the country.
Active Pharmaceutical Ingredient Regulation
The FDA will regulate finished products containing APIs developed or under development by us. Depending on its intended uses, a finished product containing the API may be regulated as a drug under the procedures described above. It may be possible to market a finished product containing an API developed or under development by us as a dietary supplement. Dietary supplements do not require FDA premarket approval. However, it may be necessary to submit a notification to the FDA that a company intends to market a dietary supplement containing a “new dietary ingredient.” In general, the regulatory requirements in other countries also depend on the nature of the finished product and do not focus on the API itself.
Recent Developments
On April 20-21, 2018, we hosted a well-attended investigators meeting for the TRILOGY Phase 3 studies in Fairfax, VA. The aim of the investigators meeting was to ensure that the clinical studies are conducted in compliance with the clinical study protocol, guidelines and applicable regulations. Approximately 200 attendees participated in this meeting which was composed of physicians, study nurses and study coordinators representing 90 of the TRILOGY clinical sites together with the clinical team of Acasti, our CRO, and the lead Principal Investigator for the TRILOGY studies, Dariush Mozaffarian, M.D., Dr.P.H., who also presented at the meeting. Dr. Mozaffarian is a highly regarded cardiologist at Tufts University, and his research focuses on the influence of omega-3s, diet and lifestyle on cardiometabolic health.
On April 24, 2018, we announced the entering into of an underwriting agreement with Mackie Research Capital Corporation (“Mackie”) in respect of a public offering of units, with each unit consisting of one common share and one common share purchase warrant (the “Offering”). On May 9, 2018, we announced the closing of the Offering pursuant to which we issued 9,530,000 units at a price of $1.05 per unit for aggregate gross proceeds to us of $10,006,500. The common share purchase warrants comprising the units are exercisable at any time prior to May 9, 2023 at an exercise price of $1.31 per common share. On May 14, 2018, we announced that Mackie had exercised the over-allotment option in full pursuant to which we issued, on the same date, 1,429,500 additional units upon the same terms as set forth above for additional aggregate gross proceeds to us of $1,500,975. In consideration for the services rendered by Mackie in connection with the Offering, we paid Mackie a cash commission equal to 7% of the gross proceeds raised under the Offering and granted non-transferrable broker warrants equal to 5% of the number of units sold under the Offering exercisable at any time prior to May 9, 2023 at an exercise price of $1.05 per common share.
On April 27, 2018, we announced the appointment of Donald Olds to our board of directors and audit committee. See “Item 6. Directors, Senior Management and Employees – Directors and Senior Management.”
On May 18, 2018, we announced that we retained Crescendo Communications, LLC to provide us with investor relations services in the United States.
On June 4, 2018, we announced the appointment of Mr. Brian Groch as our Chief Commercial Officer. Mr. Groch brings over 25 years of senior experience in the healthcare and life science industries, including product commercialization, developing and executing global sales strategies, business development, and operations. Mr. Groch will drive our global commercialization strategy including US launch planning and execution, and commercial partnering activities in the rest of the world. See “Item 6. Directors, Senior Management and Employees – Directors and Senior Management.”
Laurent Harvey, our VP of Clinical and Nonclinical Affairs, announced he will be resigning effective July 9, 2018. The TRILOGY program is well underway and enrollment is progressing according to schedule. We do not plan to replace Mr. Harvey as there is a strong clinical team in place that is well supported by our CRO and consultants.
As of June 26, 2018, we have activated 110 clinical sites, 463 patients have been enrolled and 41 patients have been randomized for the CaPre TRILOGY Phase 3 program. Additional cGMP production lots of active pharmaceutical ingredient (API) and CaPre were manufactured during the fourth quarter, enabling us to continue to accumulate the CaPre and placebo inventory required to support the TRILOGY trials.
| C. | Organizational Structure |
We have no subsidiaries. As of the date of this annual report, Neptune owns 5,064,694 of our common shares, representing 13.8% of our currently issued and outstanding common shares.
| D. | Property, Plants and Equipment |
Our head office and operations are located at 545, Promenade Centropolis, suite 100, Laval, Québec, Canada, H7T 0A3. We do not own our own manufacturing facility for the production of CaPre; however, we do own the proprietary equipment for producing the API and drug product. We currently do not have plans to develop our own manufacturing facility. However, this could change in the foreseeable future, as we consider the most cost-effective approaches to producing CaPre while ensuring the highest level of quality. We currently depend on third party suppliers and manufacturers, such as Neptune, to produce our required raw krill oil and drug substance and products. If CaPre is approved for distribution by the FDA, we initially expect to rely on cGMP-compliant third parties to manufacture NKPL66, which is API in CaPre, encapsulate, bottle and package clinical supplies of CaPre.
We have entered into an agreement CordenPharma Chenôve, a third party CMO, for the manufacturing of CaPre clinical material for the purposes of our planned TRILOGY Phase 3 program in accordance with cGMP regulations imposed by the FDA.
| - 42 - |
| Item 4A. | Unresolved Staff Comments |
Not applicable.
| Item 5. | Operating and Financial Review and Prospects |
This annual report contains forward-looking statements, principally in, but not limited to, “Item 4 - Information on the Company” and “Item 5 - Operating and Financial Review and Prospects”. These statements may be identified by the use of words like “plan”, “expect”, “aim”, believe”, “project”, “anticipate”, “intend”, “estimate”, “will”, “should”, “could” and similar expressions in connection with any discussion, expectation, or projection of future operating or financial performance, events or trends. In particular, these include statements about our strategy for growth, future performance or results of current sales and production, interest rates, foreign exchange rates, and the outcome of contingencies, such as acquisitions and/or legal proceedings and intellectual property issues.
Forward-looking statements are based on certain assumptions and expectations of future events that are subject to risks and uncertainties. Actual future results and trends may differ materially from historical results or those projected in any forward-looking statements depending on a variety of factors, including, among other things, the factors discussed in this annual report under “Item 3.D - Risk Factors” and factors described in documents that we may furnish from time to time to the SEC. Although the forward-looking information is based upon what we believe to be reasonable assumptions, no person should place undue reliance on forward-looking information since actual results may vary materially from the forward-looking information. Except as required by law, we undertake no obligation to update publicly or revise any forward- looking statements because of new information. Please refer to “Special Note Regarding Forward-Looking Statements” at the beginning of this annual report for additional details.
Management’s Discussion and Analysis of Financial Situation and Operating Results For Year Ended March 31, 2018, Thirteen-Month and One-Month Periods Ended March 31, 2017, Twelve-Month Period Ended February 28, 2017, and Year Ended February 29, 2016
Introduction
This management discussion and analysis, or MD&A, is presented in order to provide the reader with an overview of our financial results and changes to our financial position as at March 31, 2018 and for the year then ended. This MD&A also explains the material variations in our financial statements of operations, financial position and cash flows for our fiscal year ended March 31, 2018, the thirteen-month and one-month periods ended March 31, 2017, the twelve-month period ended February 28, 2017, and the fiscal year ended February 29, 2016.
This MD&A, should be read together with our audited financial statements for the fiscal year ended March 31, 2018, the thirteen-month and one-month periods ended March 31, 2017, the twelve-month period ended February 28, 2017, and the fiscal year ended February 29, 2016 under “Item 17. Financial Statements” in this annual report. Our audited financial statements were prepared in accordance with IFRS, as issued by the IASB. Our financial results are published in Canadian dollars. All amounts appearing in this MD&A are in thousands of Canadian dollars, except share and per share amounts or unless otherwise indicated.
Caution Regarding Non-IFRS Financial Measures
We use multiple financial measures for the review of our operating performance. These measures are generally IFRS financial measures, but one adjusted financial measure, Non-IFRS operating loss, is also used to assess our operating performance. This non-IFRS financial measure is derived from our financial statements and is presented in a consistent manner. We use this measure, in addition to the IFRS financial measures, for the purposes of evaluating our historical and prospective financial performance, as well as our performance relative to competitors. All of these measures also help us to plan and forecast future periods as well as to make operational and strategic decisions. We believe that providing this Non-IFRS information to investors, in addition to IFRS measures, allows them to see our results through the eyes of our management, and to better understand our historical and future financial performance.
Securities regulations require that companies caution readers that earnings and other measures adjusted to a basis other than IFRS do not have standardized meanings and are unlikely to be comparable to similar measures used by other companies. Accordingly, they should not be considered in isolation. We use Non-IFRS operating loss to measure our performance from one period to the next without the variation caused by certain adjustments that could potentially distort the analysis of trends in our operating performance, and because we believe it provides meaningful information on our financial condition and operating results. Our method for calculating Non-IFRS operating loss may differ from that used by other corporations.
| - 43 - |
We calculate our Non-IFRS operating loss measurement by adding to net loss, finance expenses, depreciation and amortization and impairment loss, change in fair value of derivative warrant liabilities, stock-based compensation and by subtracting finance income and deferred tax recovery. Other items that do not impact our core operating performance are excluded from the calculation as they may vary significantly from one period to another. Finance income/costs include foreign exchange gain (loss). We also exclude the effects of certain non-monetary transactions recorded, such as stock-based compensation, from our Non-IFRS operating loss calculation. Excluding this item does not imply it is necessarily non-recurring. A reconciliation of net loss to Non-IFRS operating loss is presented further below.
Basis of Presentation of the Financial Statements
Beginning in fiscal 2017, our fiscal year end is on March 31. Previously, our fiscal year end was February 28. Based on this change and as permitted in the transitional year by the Canadian Securities regulator, the financial statements and corresponding notes to the financial statements relating to this MD&A include for comparison purposes, thirteen months of operations, beginning on March 1, 2016 and ending on March 31, 2017 and two unaudited periods: the one-month period ended March 31, 2017 and the twelve-month period ended February 28, 2017.
Following the change of year end to March 31, 2017 for fiscal 2017 and the inclusion of thirteen months of operations, the MD&A discusses and compares the year ended March 31, 2018 to the thirteen-month and one-month periods ended March 31, 2017 and year ended February 29, 2016. In addition, there is comparative discussion of our results of operations for the three-month periods ended March 31, 2018 and February 28, 2017 and a discussion on notable items related to the one-month result of operations ending March 31, 2017. The selected quarterly financial data includes the eight most recent fiscal quarters.
We are subject to a number of risks associated with the conduct of our TRILOGY Phase 3 clinical program and its results, the establishment of strategic partnerships and the successful development of CaPre and other new products and their commercialization. We are currently not generating any revenues and have incurred significant operating losses and negative cash flows from operations since inception. To date, we have financed our operations through the public offering and private placement of Common Shares, units consisting of Common Shares and warrants and convertible debt, proceeds from research grants and research tax credits, and exercises of warrants, rights, and options. To achieve the objectives of our business plan, we plan to raise the necessary funds through additional securities offerings and the establishment of strategic partnerships as well as additional research grants and research tax credits. CaPre and other drug product candidates developed by us will require approval from the FDA and equivalent regulatory organizations in other countries before they can be commercialized. Our ability to achieve profitable operations is dependent on a number of factors outside of our control. See “Risk Factors” in this Annual Report on Form 20-F and in the SEDAR-filed MD&A for the fiscal year ended March 31, 2018.
We have incurred operating losses and negative cash flows from operations since inception. Our current assets of $9.5 million as at March 31, 2018 include cash and cash equivalents totaling $8.2 million, mainly generated by the net proceeds from the Public Offering completed on December 27, 2017. Our current liabilities total $6.7 million at March 31, 2018 and are comprised primarily of amounts due to or accrued for creditors. Since our March 31, 2018 year end, our current assets, have been increased by approximately $10.0 million from the net proceeds, of a public financing completed in early May 2018 including the exercise of the overallotment option (note 24 – subsequent event). However, in spite of this incremental financing, these current assets are projected to be significantly less than what will be needed to support the current liabilities date when combined with the projected level of expenses for the next twelve months, including the continued advancement of the TRILOGY Phase 3 clinical study program for its drug candidate, CaPre. Additional funds will also be needed for the expected expenses for the total CaPre Phase 3 research and development phase beyond the next twelve months, including the potential regulatory (NDA) submission. We also expect to incur increased general and administrative expenses (“G&A”) as a result of a planned increase in business development and commercialization planning expenses, and a reduction of its shared services agreement with Neptune, with those added expenses having begun during the year ended March 31, 2018. In addition to the recently raised additional funds, we are working toward development of strategic partner relationships and plan to raise additional funds in the future, but there can be no assurance as to when or whether we will complete any additional financing or strategic collaborations. In particular, raising financing is subject to market conditions and is not within our control. If we do not raise additional funds, find one or more strategic partners, we may not be able to realize our assets and discharge our liabilities in the normal course of business. As a result, there exists a material uncertainty that casts substantial doubt about our ability to continue as a going concern and, therefore, realize our assets and discharge our liabilities in the normal course of business. We currently have no other arranged sources of financing.
Our financial statements have been prepared on a going concern basis, which assumes we will continue our operations in the foreseeable future and will be able to realize our assets and discharge our liabilities and commitments in the ordinary course of business. These financial statements do not include any adjustments to the carrying values and classification of assets and liabilities and reported expenses that may be necessary if the going concern basis was not appropriate for these financial statements. If we were unable to continue as a going concern, material write-downs to the carrying values of the Corporation’s assets, including the intangible asset, could be required.
| - 44 - |
SELECTED FINANCIAL INFORMATION
| Three-month period ended |
One-month ended |
Three-month period ended |
Year ended |
Thirteen-month period ended |
Year ended |
|||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
February 28, 2017 |
March 31, 2018 |
March 31, 2017 |
February 29, 2016 |
|||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Net loss | (8,140 | ) | (769 | ) | (2,597 | ) | (21,504 | ) | (11,247 | ) | (6,317 | ) | ||||||||||||
| Basic and diluted loss per share | (0.32 | ) | (0.05 | ) | (0.23 | ) | (1.23 | ) | (1.01 | ) | (0.59 | ) | ||||||||||||
| Non-IFRS operating loss1 | (6,427 | ) | (406 | ) | (1,745 | ) | (16,095 | ) | (7,798 | ) | (6,569 | ) | ||||||||||||
| Total assets | 22,959 | 25,456 | 26,367 | 22,959 | 25,456 | 28,517 | ||||||||||||||||||
| Working capital2 | 2,795 | 8,143 | 8,604 | 2,795 | 8,143 | 10,184 | ||||||||||||||||||
| Total non-current financial liabilities | 8,038 | 1,615 | 1,576 | 8,038 | 1,615 | 156 | ||||||||||||||||||
| Total equity | 8,224 | 21,703 | 22,386 | 8,224 | 21,703 | 27,220 |
COMMENTS ON THE SIGNIFICANT VARIATIONS OF RESULTS FROM OPERATIONS FOR THE TWELVE-MONTH AND THE THREE-MONTH PERIODS ENDED MARCH 31, 2018 AGAINST THE THIRTEEN-MONTH AND ONE-MONTH PERIODS ENDED MARCH 31, 2017, THE THREE-MONTH PERIOD ENDED FEBRUARY 28, 2017 AND THE YEAR ENDED FEBRUARY 29, 2016
The net loss totaling $8,140 or ($0.32) per share for the three-month period ended March 31, 2018 increased by $5,543 or ($0.09) per share from the net loss totaling $2,597 or ($0.23) per share for the three-month period ended February 28, 2017. This resulted primarily from the $4,682 increased Non-IFRS operating loss, a $666 increase in loss due to the change in value of the warrant derivative liability (see “Reconciliation of Net Loss to Non-IFRS Operating Loss”), a $110 increase in stock-based compensation and a decrease of $129 of deferred tax recovery offset by a $43 decrease in financial expense.
The net loss totaling $21,504 or ($1.23) per share for the year ended March 31, 2018 increased by $10,257 or ($0.22) per share from the net loss totaling $11,247 or ($1.01) per share for the thirteen-month period ended March 31, 2017. This resulted primarily from the $8,297 increased Non-IFRS operating loss, a $1,351 increase in financial expense (see “Reconciliation of Net Loss to Non-IFRS Operating Loss”), a $291 increase in loss due to the change in value of the warrant derivative liability and a $255 increase in stock-based compensation, and a decrease of $129 in deferred tax recovery offset by a $66 decrease in depreciation and amortization.
The net loss totaling $11,247 or ($1.01) per share for the thirteen-month period ended March 31, 2017 increased $4,930 or ($0.42) per share compared to the net loss totaling $6,317 or ($0.59) per share for the year ended February 29, 2016. This change resulted primarily based on the $1,229 increased Non-IFRS operating loss explained below, $2,254 from the increased loss due to the change in value of the warrant derivative liability due to the reduction in our share price, a $1,207 financial expense increase (led by a foreign exchange gain during the prior period transitioning to a foreign exchange loss during the current period), and increased depreciation and stock compensation expense offset by no impairment charge in the current period compared to the $339 charge in the prior period combined with the $129 tax benefit recognized in the current period.
___________________________________
1The Non-IFRS operating loss (adding to net loss financial expenses (income), depreciation and amortization, change in fair value of derivative warrant liabilities and stock-based compensation) is not a standard measure endorsed by IFRS requirements. A reconciliation to the Corporation’s net loss is presented below.
2The working capital is presented for information purposes only and represents a measurement of the Corporation’s short-term financial health. The working capital is calculated by subtracting current liabilities from current assets. Because there is no standard method endorsed by IFRS requirements, the results may not be comparable to similar measurements presented by other public companies.
| - 45 - |
Breakdown of major components of the statement of earnings and comprehensive loss
| Research and development expenses | ||||||||||||||||||||||||
| Three-month period ended |
One-month ended |
Three-month period ended |
Year ended |
Thirteen-month period ended |
Year ended |
|||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
February 28, 2017 |
March 31, 2018 |
March 31, 2017 |
February 29, 2016 |
|||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Salaries and benefits | 615 | 104 | 376 | 1,705 | 1,294 | 989 | ||||||||||||||||||
| Stock-based compensation | 91 | 18 | 27 | 308 | 107 | 53 | ||||||||||||||||||
| Research contracts | 4,719 | 63 | 435 | 9,381 | 3,148 | 2,730 | ||||||||||||||||||
| Professional fees | 248 | 57 | 238 | 1,790 | 635 | 1,171 | ||||||||||||||||||
| Depreciation and amortization | 667 | 226 | 668 | 2,672 | 2,738 | 2,395 | ||||||||||||||||||
| Impairment of intangible assets | - | - | - | - | - | 339 | ||||||||||||||||||
| Other | 38 | 3 | 28 | 222 | 60 | 238 | ||||||||||||||||||
| Government grants and tax credits | (325 | ) | (45 | ) | (215 | ) | (409 | ) | (329 | ) | (349 | ) | ||||||||||||
| Total | 6,053 | 426 | 1,557 | 15,669 | 7,653 | 7,566 | ||||||||||||||||||
| General and administrative expenses | ||||||||||||||||||||||||
| Three-month period ended |
One-month ended |
Three-month period ended |
Year ended |
Thirteen-month period ended |
Year ended |
|||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
February 28, 2017 |
March 31, 2018 |
March 31, 2017 |
February 29, 2016 |
|||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Salaries and benefits | 584 | 110 | 493 | 1,576 | 1,197 | 409 | ||||||||||||||||||
| Administrative fees | 14 | 25 | 75 | 121 | 325 | 579 | ||||||||||||||||||
| Stock-based compensation | 177 | 68 | 131 | 621 | 567 | 256 | ||||||||||||||||||
| Professional fees | 428 | 52 | 231 | 1,347 | 1,049 | 616 | ||||||||||||||||||
| Other | 106 | 37 | 84 | 362 | 419 | 186 | ||||||||||||||||||
| Total | 1,309 | 292 | 1,014 | 4,027 | 3,557 | 2,046 | ||||||||||||||||||
Three-month period ended March 31, 2018 compared to the three-month period ended February 28, 2017 and the one-month period ended March 31, 2017:
During the three-month period ended March 31, 2018, we continued our planned advancement of the two study TRILOGY Phase 3 clinical study program for its drug candidate, CaPre, in partnership with one of the world’s largest providers of biopharmaceutical development and commercial outsourcing services (“CRO”). The $6,053 in total R&D expenses for the three-month period ended March 31, 2018 totaled $5,295 before depreciation, amortization and stock-based compensation expense, compared to $1,557 in total R&D expenses for the three-month period ended February 28, 2017 or $862 before depreciation, amortization and stock-based compensation expense. This $4,433 increase in R&D expenses before depreciation, amortization and stock-based compensation was mainly attributable to the $4,284 increase in research contracts, $239 increase in salaries and benefits and an increase of $110 related to tax credits. The increased research contract expense resulted primarily from a planned $3,277 increase in the CRO Phase 3 clinical trial program contract expense with continued site activation and patient enrollment and treatment and an amount of $992 of increased research contracts resulting from the planned expanded scale-up production activities relating to CaPre during the three-month period ended March 31, 2018 compared to the three-month period ended February 28, 2017. An increase of $239 in incremental salaries and benefits primarily related to full-time leadership and management of CMC regulatory affairs in R&D combined with the addition of several technicians to production and quality control earlier in the current fiscal year when compared to the three-month period ended February 28, 2017. The $110 increase in tax credits relates to higher R&D expenditures combined with a higher investment tax credit rate in the three-month period ending March 31, 2018.
G&A expenses totaling $1,132 before stock-based compensation expense for the three-month period ending March 31, 2018 increased by $249 from $883 for the three-month period ended February 28, 2017. This $249 increase was mainly attributable to a $91 increase in salaries and benefits associated with adding full-time executive and managerial headcount to support our strategy and financing while becoming more independent from Neptune, partially offset by a $61 reduction in Neptune administrative fees and an increase in professional fees of $197. The professional fee increase was due primarily to additional legal fees resulting from increased independence from Neptune, including no continued internal counsel services, and the further building of our reactivated public and investor relations program.
| - 46 - |
Year ended March 31, 2018 compared to the Thirteen-month and one-month periods ended March 31, 2017:
As we continued advancing our planned Phase 3 clinical program and production scale-up of CaPre within its R&D program, $15,669 was incurred in total R&D expenses for the year ended March 31, 2018 and $12,689 was incurred before depreciation, amortization and stock-based compensation expense. This compares to $7,653 in total R&D expenses for the thirteen-month period ended March 31, 2017 or $4,808 before depreciation, amortization and stock-based compensation expense. This 7,881 increase in R&D expenses before depreciation, amortization and stock-based compensation was mainly attributable to the $6,233 increase in contracts with a $5,858 increase in Phase 3 CRO contract expenses offset by a $1,663 decrease in PK Bridging and other clinical study program contract expenses incurred during the prior-year thirteen-month period, and a $2,038 increase in contract manufacturing (“CMO”) production expenses. There was also a $1,155 increase in professional fees primarily incurred in completing due diligence and preliminary discussions for strategic R&D partnership and licensing arrangements. Salary and benefits additionally contributed to the overall increase by $411 related to R&D management combined with additional headcount for production and quality control as the Corporation advanced its Phase 3 clinical study program. The $80 increase to tax credits relates mainly to a higher investment tax credit rate combined with increased R&D expenditures in the year ended March 31, 2018 compared to the thirteen-month period ended March 31, 2017.
G&A expenses totaling $3,406 before stock-based compensation expense for the year ended March 31, 2018 increased by $416 from $2,990 for the thirteen-month period ended March 31, 2017. This $416 increase was mainly attributable to a $379 increase in salaries and benefits associated with adding full-time executive and managerial headcount to support our strategy and financing while becoming more independent from Neptune, offset by a $204 reduction in Neptune administrative fees. This increase also resulted from increased professional fees of $298 due primarily to additional legal fees resulting from increased independence from Neptune and expenses relating to further building our reactivated public and investor relations programs, as well as a decrease of $57 in other expenses.
Thirteen-month and one-month periods ended March 31, 2017 compared to the year-ended February 29, 2016:
R&D expenses totaled $7,653 for the thirteen-month period ended March 31, 2017 or an increase of $87 compared to $7,566 in total R&D expenses for the year ended February 29, 2016. The R&D expense increase resulted primarily from $426 in total R&D expenses for March 2017, the thirteenth month of the current period ended March 31, 2017, offset by no intangible asset impairment charge in this period ended March 31, 2017 compared to the $339 charge during fiscal 2016. R&D expenses, before consideration of stock-based compensation, amortization and depreciation and impairments of intangible assets, increased by $29 for the thirteen-month period ended March 31, 2017, including $182 for the month of March 2017, to total $4,808 compared to $4,779 for the year ended February 29, 2016. The increase of $29 was mainly attributable to the increase in research contracts of $419 and salaries and benefits of $305, principally offset by decreases in professional fees of $537, other expenses of $177 and government grants of $19. The current period’s increase of $419 in research contracts includes $63 relating to the additional one-month period ended March 31, 2017, but was primarily due to the cost of the Phase 2 bioavailability bridging clinical study initiated early in fiscal 2017 exceeding the cost of the other Phase 2 and nonclinical testing completed in fiscal 2016. The increased salaries and benefits represented the cost of the expanded team headcount, led by full-time dedicated management (only part time in prior years), needed for the Corporation to continue its pharmaceutical process and analytical development and chemistry manufacturing control scale-up, as planned on our previously announced timeline. The decrease of $537 in professional fees is primarily due to a decrease in the development consulting fees incurred in fiscal 2016 for the prior Phase 2 clinical study analytics and the planning for the Phase 2 bridging clinical study.
G&A expenses totaled $3,557 for the thirteen-month period ended March 31, 2017 or an increase of $1,511 compared to total G&A expenses of $2,046 for the year ended February 29, 2016. This period-to-period increase includes $292 in total G&A expenses for the thirteenth month of March 2017, $243 in increased stock-based compensation expense and a $976 increase in other G&A expenses, excluding the thirteenth month and stock-based compensation expenses. G&A expenses, excluding the stock-based compensation, increased $1,200 to $2,990 for the thirteen-month period ended March 31, 2017, including $224 for the month of March 2017, compared to $1,790 for the year ended February 29, 2016. This increase was primarily attributable to a $788 increase in salaries and benefits offset by a $254 decrease in Neptune administrative fees, combined with increased professional fees of $433, and other expenses of $233. The increase in salaries and benefit expenses resulted from our need for the added full-time executive and managerial headcount to lead our strategy, incremental financing and back office while supporting continued and expanded R&D with the need for full-time leadership from its management (which was only part time in prior years). The increased professional fees were principally comprised of expenses associated with the investor and public relations program, the achievement of business development milestones, increased market research expenses, and non-recurring project legal and accounting fees associated with the year-end change and the immigration-related fees for the U.S.-resident executives.
| - 47 - |
RECONCILIATION OF NET LOSS TO NON-IFRS OPERATING LOSS
| Three-month period ended |
One-month ended |
Three-month period ended |
Year ended |
Thirteen-month period ended |
Year ended |
|||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
February 28, 2017 |
March 31, 2018 |
March 31, 2017 |
February 29, 2016 |
|||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Net loss | (8,140 | ) | (769 | ) | (2,597 | ) | (21,504 | ) | (11,247 | ) | (6,317 | ) | ||||||||||||
| Add (deduct): | ||||||||||||||||||||||||
| Stock-based compensation | 268 | 86 | 158 | 929 | 674 | 309 | ||||||||||||||||||
| Depreciation and amortization | 667 | 226 | 668 | 2,672 | 2,738 | 2,395 | ||||||||||||||||||
| Impairment of intangible assets | - | - | - | - | - | 339 | ||||||||||||||||||
| Financial expenses (income) | (15 | ) | 29 | 28 | 1,464 | 113 | (1,094 | ) | ||||||||||||||||
| Change in fair value of | ||||||||||||||||||||||||
| Derivative warrant liabilities | 793 | 22 | 127 | 344 | 53 | (2,201 | ) | |||||||||||||||||
| Deferred income tax Recovery | - | - | (129 | ) | - | (129 | ) | - | ||||||||||||||||
| Non-IFRS operating loss | (6,427 | ) | (406 | ) | (1,745 | ) | (16,095 | ) | (7,798 | ) | (6,569 | ) |
Stock-based compensation expense increased by $110 to $268 for the three-month period ended March 31, 2018 from $158 for the three-month period ended February 28, 2017. No options were granted in the three-month period ending March 31, 2018 nor in the three-month period ending February 29, 2017.
Stock-based compensation expense increased by $255 to $929 for the year ended March 31, 2018 from $674 for the thirteen-month period ended March 31, 2017. There was a decrease of 178,900 options granted in the year ended March 31, 2018 compared to the thirteen-month period ended March 31, 2017. The increase in stock-based compensation resulted primarily from the number of options vesting in the comparable periods. At March 31, 2018, 591,113 options were fully vested and exercisable compared to 238,482 at March 31, 2017. The overall stock-based compensation expense increased for the thirteen-month period ending March 31, 2017 as a total of 1,300,400 stock options were granted compared to 109,188 stock options being granted for the year ended February 29, 2016.
The depreciation and amortization expense decreased by $1 to $667 for the three-month period ended March 31, 2018 from $668 for the three-month period ended February 28, 2017, remaining constant. The depreciation and amortization expense decreased on a net basis by $66 to $2,672 for the twelve-month period ended March 31, 2018 from $2,738 for the thirteen-month period ended March 31, 2017, due to increased depreciation for the current year’s production equipment additions being partially offset by the reduction to twelve months in the current year. Depreciation and amortization expense totaled $2,738 for the thirteen-month period ended March 31, 2017 which approximated the same amount when compared to the year ended February 29, 2016, when reduced by the extra month for the period ended March 31, 2017. The $339 impairment charge was recognized only during the year ended February 29, 2016.
Financial expenses decreased by $43 to financial income of $15 for the three-month period ended March 31, 2018 from financial expenses of $28 for the three-month period ended February 28, 2017. This resulted primarily from an increase in interest revenue of $30 to $33 for the three-month period ended March 31, 2018 from $3 for the three-month period ended February 28, 2017. Additionally, the change resulted from a $127 increase in foreign exchange gain from a loss of $22 for the three-month period ended February 28, 2017 to a gain of $105 for the three-month period ended March 31, 2018. An increase of $33 expenses related to financing transaction costs occurred, with costs incurred of $33 for the three-month period ended March 31, 2018 from nil for the three-month period end February 28, 2017. This change was offset by the increase in interest expense on convertible debentures of $83 for the three-month period ended March 31, 2018 amounting to $91 compared to $8 for the three-month period ended February 28, 2017, and a decrease of $2 in other charges for the three-month period ended March 31, 2018 compared to the three-month period ended February 28, 2017.
Financial expenses increased by $1,351 to $1,464 for the year ended March 31, 2018 from $113 for the thirteen-month period ended March 31, 2017. This resulted primarily from transaction costs totaling $1,134 for the year ended March 31, 2018 compared to nil for the thirteen-month period ended March 31, 2017. This changed also from a reduction of interest income of $53 to $72 for the year ended March 31, 2018 from $125 for the thirteen-month period ended March 31, 2017. Additionally, the change was offset by a $148 reduced foreign exchange loss from a loss of $180 for the thirteen-month period ended March 31, 2017 to a loss of $32 for the year ended March 31, 2018. This change also resulted from an increase in interest expense on convertible debentures of $327 for the year ended March 31, 2018 compared to $39 for the thirteen-month period ended March 31, 2017, and a decrease of $15 in other charges to the thirteen-month period ended March 31, 2017.
| - 48 - |
Net financial expenses (income) totaling $113 for the thirteen-month period ended March 31, 2017 reflect a $1,207 decrease compared to ($1,094) for the year ended February 29, 2016 primarily resulting from the $1,023 foreign exchange gain recognized during the year ended February 29, 2016 changing to the $180 foreign exchange loss recognized during the thirteen-month period ended March 31, 2017. The foreign exchange changes resulted primarily from the utilization of US$-denominated cash and cash equivalents over the periods generating lower US-denominated cash and cash equivalents throughout the periods and at March 31, 2017 compared to February 29, 2016 and, the periods then ended combined with a decrease in the reporting US exchange rate. The US$-denominated cash, cash equivalents and short-term investments totaled US$3,524 at March 31, 2017 and US$10,314 at February 29, 2016 and the exchange rate reporting of CA$ per US$ was $1.3299 at March 31, 2017 compared to $1.3531 at February 29, 2016. Additionally, interest income for the current thirteen-month period totaled $125 compared to $73 for the year ended February 29, 2016, and $39 in interest expense was incurred in the current period, including $31 in March, in association with the convertible debentures from the Private Placement.
The fair value of the derivative warrants issued with the U.S. Public offering of December 27, 2017 was determined to be $0.60 per warrant and totaled $5,873 upon issuance. The fair value of the warrants is re-measured at each reporting date using the Black-Scholes option pricing model. At March 31, 2018, the fair value of these warrants totaled $6,405 or $0.65 per warrant. The change in our stock price and the FX conversion resulted in a loss of $532 on the fair value of the warrants increasing the corresponding liability.
The fair value of the derivative warrant liabilities issued in December 2013 totaled $21 at March 31, 2018 or $188 less than the $209 fair value at March 31, 2017 and $22 less than the $187 fair value at February 28, 2017. The fair value of the warrants is estimated at each reporting date using the Black-Scholes option pricing model. The fair value of the warrants issued in connection with our previous securities offerings was determined to be $0.01 per warrant upon issuance, $0.01 per warrant at March 31, 2018, $0.11 per warrant at March 31, 2017 and $0.10 per warrant at February 28, 2017. During the three-month period and year ended March 31, 2018, the fluctuation in our stock price, the overall decline in the FX conversion rate and the reduction of the estimated life of the warrants resulted in a gain on the change in fair value of the warrant liabilities reducing the corresponding liability in the statement of financial position. The fair value of the derivative warrant liabilities totaled $209 at March 31, 2017 or $53 more than the $156 fair value at February 29, 2016, $22 of which was recognized during the one-month ended March 31, 2017.
We recorded a $129 deferred income tax recovery at February 28, 2017 to reduce to nil an income tax liability that was attributable to the difference between the tax basis and the carrying amount of the unsecured convertible debentures.
Non-IFRS operating loss increased by $4,682 for the three-month period ended March 31, 2018 to $6,427 compared to $1,745 for the three-month period ended February 28, 2017. This was primarily due to an increase in research and development (“R&D”) expenses of $4,433 and an increase in G&A expenses of $249, before consideration of stock-based compensation, amortization and depreciation. Non-IFRS operating loss increased by $8,297 for the year ended March 31, 2018 to $16,095 compared to $7,798 for the thirteen-month period ended March 31, 2017. This primarily resulted due to an increase in R&D expenses of $7,881 and an increase in G&A expenses of $416, before consideration of stock-based compensation, amortization and depreciation. The Non-IFRS operating loss increased by $1,229 for the thirteen-month period ended March 31, 2017 to $7,798 compared to $6,569 for the year-ended February 29, 2016. This increase was primarily due to the incremental one-month period Non-IFRS operating loss of $406 for March 2017 as well as increased G&A expenses compared to the prior period before consideration of stock-based compensation and amortization and depreciation.
SELECTED QUARTERLY FINANCIAL DATA
| March 31, | December 31, | September 30, | June 30, | |||||||||||||
| 2018 | 2017 | 2017 | 2017 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (8,140 | ) | (6,079 | ) | (4,507 | ) | (2,778 | ) | ||||||||
| Add (deduct): | ||||||||||||||||
| Depreciation and amortization | 667 | 671 | 667 | 667 | ||||||||||||
| Stock based compensation | 268 | 330 | 295 | 36 | ||||||||||||
| Financial expenses (income) | (15 | ) | 1,220 | 146 | 113 | |||||||||||
| Change in fair value of derivative warrant liabilities | 793 | (291 | ) | (24 | ) | (134 | ) | |||||||||
| Non-IFRS operating loss | (6,427 | ) | (4,149 | ) | (3,423 | ) | (2,096 | ) | ||||||||
| Basic and diluted net loss per share | (0.32 | ) | (0.40 | ) | (0.31 | ) | (0.19 | ) |
| - 49 - |
| March 31, | November 30, | August 31, | May 31, | |||||||||||||
| 20173 | 2016 | 2016 | 2016 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Net loss | (3,366 | ) | (2,397 | ) | (2,329 | ) | (3,155 | ) | ||||||||
| Add (deduct): | ||||||||||||||||
| Depreciation and amortization | 894 | 621 | 614 | 609 | ||||||||||||
| Stock based compensation | 244 | 155 | 211 | 64 | ||||||||||||
| Financial expenses (income) | 57 | (117 | ) | (55 | ) | 228 | ||||||||||
| Change in fair value of derivative warrant liabilities | 149 | 2 | (66 | ) | (32 | ) | ||||||||||
| Deferred income tax recovery | (129 | ) | - | - | - | |||||||||||
| Non-IFRS operating loss | (2,151 | ) | (1,736 | ) | (1,625 | ) | (2,286 | ) | ||||||||
| Basic and diluted net loss per share | (0.28 | ) | (0.22 | ) | (0.22 | ) | (0.29 | ) |
The quarterly year-to-year non-IFRS operating loss variances are mainly attributable to fluctuations in R&D expenses from quarter-to-quarter as well as an increase in G&A expenses over the last four quarters. The increase in net loss, net loss per share and non-IFRS operating loss in the fourth quarter of 2018 can primarily be explained by the costs incurred in CRO expenses associated with its Phase 3 clinical trial program. The variances in net loss from quarter to quarter are mainly due to the changes in fair value of the warrant liabilities as well as variations in foreign exchange gains or losses.
LIQUIDITY AND CAPITAL RESOURCES
Share Capital Structure
Our authorized share capital consists of an unlimited number of Class A, Class B, Class C, Class D and Class E shares, without par value. Issued and outstanding fully paid shares, stock options, restricted shares units and warrants, were as follows for the periods ended:
| March 31, 2018 |
March 31, 2017 |
February 29, 2016 |
||||||||||
| Number outstanding |
Number outstanding |
Number outstanding |
||||||||||
| Class A shares, voting, participating and without par value | 25,638,215 | 14,702,556 | 10,712,038 | |||||||||
| Stock options granted and outstanding | 2,284,388 | 1,424,788 | 454,151 | |||||||||
| December 2017 U.S. public offering of warrants exercisable at US$1.26, until December 27, 2022 | 9,802,935 | - | - | |||||||||
| Series December 2017 U.S. Broker warrants exercisable at US$1.2625, until December 27, 2022 | 495,050 | - | - | |||||||||
| February 2017 public offering of warrants exercisable at $2.15, until February 21, 2022 | 1,904,034 | 1,965,259 | - | |||||||||
| Series February 2017 BW Broker warrants exercisable at $2.15, until February 21, 2018 | - | 234,992 | - | |||||||||
| Series 2017 unsecured convertible debentures conversion option contingent warrants exercisable at $1.90, until February 21, 20204 | 1,052,630 | 1,052,630 | - | |||||||||
| Series 8 warrants exercisable at US$15.00, until December 3, 20185 | 1,840,000 | 1,840,000 | 1,840,000 | |||||||||
| Series 9 warrants exercisable at $13.30 until December 3, 2018 | 161,654 | 161,654 | 161,654 | |||||||||
| Total fully diluted shares | 43,178,906 | 21, 381,879 | 13,167,843 |
__________________________
3 This fiscal quarter represents a period of four months ended March 31, 2017.
4 The debentures are convertible into Common Shares at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures. Should the Corporation pay all or any portion of the convertible debenture before maturity, then warrants become exercisable at $1.90 per Common Share for the equivalent convertible debenture amount prepaid.
5 Total of 18,400,000 warrants. In order to obtain one Common Share, 10 warrants must be exercised for a total amount of US$15.00
| - 50 - |
Comparison of cash flows and financial condition for the three and twelve-month periods ended March 31, 2018 and the one-month period ended March 31, 2017, three-month periods ended February 28, 2017 and thirteen-month period ended March 31, 2017 and years ended February 29, 2016
Summary
As at March 31, 2018, cash and cash equivalents totaled $8,223, with a net source of cash totaling $4,252 for the three-month period and a use of cash of $1,549 for the year ended March 31, 2018. This compares to $9,772 in total cash and cash equivalents as at March 31, 2017, with a net source of cash totaling $6,745 for the thirteen-month period and $7,546 for the twelve-month period ended February 28, 2017 with a use of cash totaling $801 for the month ended March 31, 2017. Our cash increased by $1,716 for the year ended February 29, 2016.
Operating activities
During the three-month periods ended March 31, 2018 and February 28, 2017, our operating activities used cash of $4,249 and $1,425, respectively, and during the year ended March 31, 2018 and the thirteen-month period ended March 31, 2017, our operating activities used cash of $12,519 and $6,958, respectively, further modified by changes in working capital, excluding cash. The use of cash flows in operating activities for the three-month periods ended March 31, 2018 and February 28, 2017 and the year ended March 31, 2018 and thirteen-months periods ended March 31, 2017 when compared to the net losses for each period are mainly attributable to the change in non-cash expenses, (see “Reconciliation of Net Loss to Non-IFRS Operating Loss”), further modified by changes in working capital, excluding cash.
During the year ended February 29, 2016, our operating activities used cash of $6,574 as primarily explained in the Non-IFRS operating loss section above. The use of cash flows in operating activities for the year ended February 29, 2016 when compared to the net losses for the period is mainly attributable to the change in non-cash operating items, as explained in the Reconciliation of Net Loss to Non-IFRS Operation Loss section above offset by reductions in working capital, excluding cash.
Investing activities
During the three-month period ended March 31, 2018, our investing activities used cash of $236 compared to generating cash of $3,327 for the three-month period ended February 28, 2017. Cash used by investing activities during the three-month period ended March 31, 2018 was due to the acquisition of equipment of $128, acquisition of marketable securities of $26, offset by interest received of $31. Cash generated by investing activities for the three-month period ended February 28, 2017 was mainly due to the maturity of short-term investments of $4,031, partially offset by the acquisition of equipment totaling $733.
During the year ended March 31, 2018, our investing activities used cash of $411 compared to generating cash of $6,888 for the thirteen-month period ended March 31, 2017. Cash used by investing activities during the year ended March 31, 2018 was due to the acquisition of equipment totaling $455, acquisition of marketable securities of $26, partially offset by interest received of $70. Cash generated by investing activities for the thirteen-month period ended March 31, 2017 was mainly due to the maturity of short-term investments of $22,030, partially offset by a $12,765 reinvestment in short-term investments and the acquisition of equipment totaling $2,527.
During the year ended February 29, 2016, our investing activities generated cash of $8,229. The cash generated by investing activities during the year-ended February 29, 2016 was mainly due to the maturity of short-term investments of $20,437, offset by the reinvestment in short-term investments totaling $11,954 and acquisition of equipment of $276.
Financing activities
During the three-month periods ended March 31, 2018, our financing activities used cash of $36 and for February 28, 2017 the Corporation generated cash of $6,924 primarily from the net proceeds of the public offering of $5,044 and net proceeds from Private Placement of $1,882.
During the year ended March 31, 2018, our financing activities generated cash of $11,406 primarily to the net proceeds from the public offering of $11,446. During the thirteen-month period ended March 31, 2017, our financing activities generated cash of $6,864 and were mainly due to the net proceeds from the Public Offering of $5,010 and net proceeds from the Private Placement of $1,872.
See basis of presentation for additional discussion of our financial condition, including the need for additional funds and the material uncertainty that casts substantial doubt about our ability to continue as a going concern.
| - 51 - |
December 2017 U.S. Public Offering
On December 27, 2017, we closed a public offering issuing 9,900,990 units of Acasti (“Units”) at a price of $1.28 (US$1.01) per Unit for gross proceeds of $12.6 million (US$10 million). The Units issued consisted of 9,900,990 Common Shares and 8,910,891 warrants with the right to purchase one Common Share of Acasti at an exercise price of US$1.26 or about $1.59 as of the issuance date and exercisable until December 27, 2022. As part of this closing, the underwriters also partially exercised for nil consideration the over-allotment option for warrants, which were issued with a right to purchase 892,044 Common Shares also at an exercise price of US$1.26 or about $1.59 as of the issuance date and also exercisable until December 27, 2022.
On January 22, 2018, the underwriters exercised a portion of their remaining over-allotment option by purchasing an additional 766,179 Common Shares at the same price of US$1.01 per share for additional gross proceeds of $963 (US$773).
The Warrants forming part of the Units are classified as Derivative Warrant Liabilities for accounting purposes given the currency of the warrant exercise price (US$) is different from our Canadian dollar functional currency. The proceeds of the offering are required to be split between the Derivative Warrant Liabilities and the equity-classified Common Shares at the time of issuance of the Units. The fair value of the Derivative Warrant Liabilities at the time of issuance was $5.9 million and the residual of the proceeds was allocated to the Common Shares. Issuance costs totaled approximately $2.5 million. These issuance costs have been allocated between the warrants and Common Shares based on relative value. The portion allocated to the Warrants was recognized in finance costs in the Interim Statements of Earnings and Comprehensive Loss, whereas the portion allocated to Common Shares was recognized as a reduction to share capital, in the Statements of Financial Position.
The fair value of these public offering Warrants issued was determined to be $0.60 per warrant as at December 27, 2017, $0.57 at December 31, 2017 and $0.65 at March 31, 2018. Changes in the fair value of the Warrants are recognized in finance income or costs.
As part of the issuance costs of this public offering, the Corporation also issued broker warrants to purchase up to 495,050 Common Shares. Each broker warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of US$1.2625 or about $1.60 as of the issuance date, at any time until December 27, 2022. The broker warrants are considered as compensation to non-employees under IFRS 2, stock-based compensation, and are accounted for at fair value through contributed surplus. The fair value of the Broker Warrants amounted to $406 based on the Black-Scholes pricing model and was allocated to share capital.
Financial Position
The following table details the significant changes to the statements of financial position as at March 31, 2018 compared to the prior fiscal period end at March 31, 2017:
| Accounts | Increase (Decrease) |
Comments | ||||
| Cash and cash equivalents | (1,549 | ) | See cash flow statement | |||
| Receivable | 553 | Timing of receipts | ||||
| Prepaid expenses | 103 | Completion of research contracts | ||||
| Other Asset – current and long term | 659 | Acquisition of Research Supplies | ||||
| Equipment | 34 | Acquisition of equipment and depreciation | ||||
| Intangible asset | (2,323 | ) | Amortization | |||
| Trade and other payables | 4,559 | Increased expenses and accruals | ||||
| Derivative warrant liabilities | 6,217 | Issuance of derivative warrants and change in fair value | ||||
| Unsecured convertible debentures | 206 | Accretion of interest | ||||
See the statement of changes in equity in our financial statements for details of changes to the equity accounts since March 31, 2017.
Derivative warrant liabilities
The warrants issued in connection with U.S. offerings are derivative liabilities (“Derivative Warrant Liabilities”) for accounting purposes due to the currency of the exercise price (US$) being different from our Canadian dollar functional currency. The warrant liabilities will be settled in Common Shares. The fair value of the warrants is revalued at each reporting date.
On December 27, 2017, warrants were issued as part of our U.S. public offering and recognized as Derivative Warrant Liabilities with a fair value of $5,873. As of March 31, 2018, the Derivative Warrant Liabilities totaled $6,405 which represents the fair value of these warrants. The fair value of the warrants issued in connection with the offering was determined to be $0.60 per warrant upon issuance and $0.65 per warrant as of March 31, 2018.
| - 52 - |
As of March 31, 2018, $21 included in liabilities represents the fair value of warrants issued as part of our December 2013 securities offering. The fair value of the warrants issued in connection with this offering was determined to be $0.58 per warrant upon issuance and $0.01 per warrant as of March 31, 2018.
Contractual Obligations, Off-Balance-Sheet Arrangements and Commitments
As at March 31, 2018, our liabilities total $14,735, of which $6,697 is due within twelve months, $6,426 relates to Derivative Warrant Liabilities that will be settled in Common Shares and $1,612 of outstanding unsecured convertible debentures also projected to be settled in Common Shares. However, the principal amount of unsecured convertible debentures may be prepaid, in whole or in part, at any time and from time to time, in cash, at the sole discretion of the Corporation. The debentures are convertible into Common Shares at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures.
The Corporation has also entered into a contract to purchase production equipment to be used in the manufacturing of the clinical and future commercial supply of CaPre.
A summary of the contractual obligations at March 31, 2018, is as follows:
| Carrying value | Total contractual cash flows |
1 year or less | 1 to 3 years | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Trade, other payables and due to related party | 6,697 | 6,697 | 6,697 | - | ||||||||||||
| Purchase obligation of equipment | 143 | 143 | 143 | - | ||||||||||||
| Lease |
151 |
151 |
72 |
79 |
||||||||||||
| Unsecured convertible debentures | 1,612 | 2,303 | 160 | 2,143 | ||||||||||||
| Total | 8,603 | 9,294 | 7,072 | 2,222 |
The Corporation has no off-balance sheet arrangements.
Research and development contracts and contract research organizations agreements
The Corporation utilizes CMOs related to the development of clinical materials and research organizations to perform services related to our clinical trials. Pursuant to the agreements with these contract manufacturing and contract research organizations, the Corporation has either the right to terminate the agreements without penalties or under certain penalty conditions. For agreements which contain penalty conditions, we would be required to pay penalties of approximately $172.
Lease
During the year ended March 31, 2018, the Company entered into a lease agreement, for its research and development and quality control laboratory facility located in Sherbrooke, Québec, resulting in a total commitment of $151 over the two-year lease term. An amount of $72 is committed in the next year, with a remaining committed amount of $79 over the second year of the lease.
Contingencies
A former CEO of the Corporation is claiming the payment of approximately $8.5 million and the issuance of equity instruments from the Neptune group (including Acasti). As our management believes that these claims are not valid, no provision has been recognized. The Neptune group (including Acasti) has filed a claim to recover certain amounts from the former CEO. All outstanding share-based payments held by the former CEO were cancelled during our fiscal year ended February 28, 2015.
The Corporation is also involved in other matters arising in the ordinary course of its business. Since management believes such claims are not valid and it presently is not possible to determine the outcome of these matters, no provisions have been made in the financial statements for their ultimate resolution beyond the amounts incurred and recorded for such matters. The resolution of such matters could have an effect on our financial statements in the year that a determination is made. However, in management's opinion, the final resolution of all such matters is not projected to have a material adverse effect on our financial position.
| - 53 - |
Related Party Transactions
Neptune was previously the parent of Acasti and owned approximately 34.0% prior to the December 2017 US public financing. After that financing, Neptune owned approximately 19.8% of the issued and outstanding Common Shares of the Corporation and that ownership has now been diluted to 13.8% after the Canadian public financing in May 2018.
The Corporation intends to continue to rely on the support of Neptune for a portion of its G&A needs in the near term; however, the continuance of this support is outside of our control.
The Corporation was charged by Neptune, for the purchase of research supplies and for certain costs incurred by Neptune for the benefit of the Corporation, as follows:
| Year ended | Thirteen-months ended |
Month ended | Year ended | Year ended | ||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 |
February 28, 2017 |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Research and development expenses | ||||||||||||||||||||
| Supplies and incremental costs | 7 | - | - | - | 5 | |||||||||||||||
| Shared service agreement | 20 | 60 | 1 | 59 | 366 | |||||||||||||||
| 27 | 60 | 1 | 59 | 371 | ||||||||||||||||
| General and administrative expenses | ||||||||||||||||||||
| Supplies and incremental costs | 239 | 293 | 16 | 277 | 299 | |||||||||||||||
| Shared service agreement | 121 | 325 | 25 | 300 | 491 | |||||||||||||||
| 360 | 618 | 41 | 577 | 790 | ||||||||||||||||
| 387 | 678 | 42 | 636 | 1,161 |
Where Neptune incurs specific incremental costs for the benefit of the Corporation, it charges those amounts directly. During the three-months and year ended March 31, 2018, the Corporation recognized an expense of $65 and $239, respectively, in G&A expenses and nil and $7, respectively, in R&D expenses relative to the expenses for the three-month period ended February 28, 2017 and thirteen-month period ended March 31, 2017 of $125 and $293, respectively, in G&A, and nil and nil, respectively, in R&D.
In addition, Neptune provided us with the services of personnel for certain of its administrative, legal and laboratory work as part of a shared service agreement. The employees’ salaries and benefits are charged proportionally to the time allocation agreed upon. In the three-months and year ended March 31, 2018, the Corporation recognized an expense of $15 and $121, respectively, in G&A expenses and nil and $20, respectively, in R&D expenses under the shared service agreement compared for the three-month period ended February 28, 2017 and thirteen-month period ended March 31, 2017 to $75 and $325, respectively, in G&A expenses, and $45 and $60, respectively, in R&D expenses.
As of August 31, 2017, the laboratory support, the corporate affairs and the public company reporting services previously provided by Neptune as part of the shared service agreement were discontinued. The Corporation is now incurring some incremental costs and expects to do so in the future, for being provided these services directly or through qualified third parties, partially offset by reduced shared service fees. The payable to Neptune primarily for G&A shared services has no specified maturity date for payment or reimbursement and does not bear interest.
These charges do not represent all charges incurred by Neptune that may have benefited the Corporation. Also, these charges do not necessarily represent the cost that the Corporation would otherwise need to incur, should it not receive these services or benefits through the shared resources of Neptune.
Historically, Neptune has provided the Corporation with the krill oil needed to produce CaPre for our clinical programs, including all of the krill oil projected as needed for its Phase 3 clinical study program. However, Neptune discontinued its krill oil production and sold its krill oil inventory to Aker on August 7, 2017. In the six-month period ending March 31, 2018, we purchased a reserve of krill oil amounting to a net of $918 from Aker that will be used in the production of CaPre capsules for its Phase 3 clinical trials as well as potential future commercial needs. The Corporation believes that alternative supplies of krill oil that can meet our specifications will be readily available and is currently evaluating alternative suppliers of krill oil. At March 31, 2018, a reserve of krill oil was still stored at Neptune’s facility.
On January 7, 2016 Neptune announced the acquisition of Biodroga Nutraceuticals Inc. As part of this transaction, the Corporation pledged $2 million of committed funds to partly guarantee the financing for the transaction. Neptune had agreed to pay us an annual fee on the committed funds outstanding at an annual rate of 9% during the first six months and 11% for the remaining term of the pledge agreement. On September 20, 2016, Neptune fully released the pledged amount. The Corporation recognized interest revenue in the amount of $89 during the thirteen-month period ended March 31, 2017 and nil for the month ended March 31, 2017.
| - 54 - |
The key management personnel are the officers of the Corporation and the members of the Board of Directors of the Corporation. They control in the aggregate less than 1% of the voting shares of the Corporation (2% in 2017). See note 6 to the financial statements for disclosures of key management personnel compensation.
Use of estimates and measurement of uncertainty
The preparation of the financial statements in conformity with IFRS requires management to make judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets, liabilities, income and expenses. Actual results may differ from these estimates.
Estimates are based on management’s best knowledge of current events and actions that the Corporation may undertake in the future. Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected.
Critical judgments in applying accounting policies that have the most significant effect on the amounts recognized in the financial statements include the following:
| · | Identification of triggering events indicating that the intangible assets might be impaired. |
| · | The use of the going concern basis of preparation of the financial statements. At the end of each reporting period, management assesses the basis of preparation of the financial statements. The financial statements have been prepared on a going concern basis in accordance with IFRS. The going concern basis of presentation assumes that the Corporation will continue its operations for the foreseeable future and can realize its assets and discharge its liabilities and commitments in the normal course of business. |
Assumptions and estimation uncertainties that have a significant risk of resulting in a material adjustment within the next financial year include the following:
| · | Determination of the recoverable amount of our cash generating unit (“CGU”). |
| · | Measurement of derivative warrant liabilities and stock-based compensation. |
Also, management uses judgment to determine which research and development (“R&D”) expenses qualify for R&D tax credits and in what amounts. The Corporation recognizes the tax credits once it has reasonable assurance that they will be realized. Recorded tax credits are subject to review and approval by tax authorities and therefore, could be different from the amounts recorded.
Critical Accounting Policies
Impairment of non-financial assets
The carrying value of our license asset is reviewed at each reporting date to determine whether there is any indication of impairment. If any such indication exists, then the CGU’s recoverable amount is estimated. The identification of impairment indicators and the estimation of recoverable amounts require the use of judgment.
Derivative warrant liabilities
The warrants forming part of the Units issued from the 2017 and 2014 public offering are derivative liabilities for accounting purposes due to the currency of the exercise price being different from our functional currency. The derivative warrant liabilities are required to be measured at fair value at each reporting date with changes in fair value recognized in earnings. Our uses Black-Scholes pricing model to determine the fair value. The model requires the assumption of future stock price volatility, which is estimated based on weighted average historic volatility. Changes to the expected volatility could cause significant variations in the estimated fair value of the derivative warrant liabilities.
Stock-based compensation
The Corporation has a stock-based compensation plan, which is described in note 16 of the financial statements. The Corporation accounts for stock options granted to employees based on the fair value method, with fair value determined using the Black-Scholes model. The Black Scholes model requires certain assumptions such as future stock price volatility and expected life of the instrument. Expected volatility is estimated based on weighted average historic volatility. The expected life of the instrument is estimated based on historical experience and general holder behavior. Under the fair value method, compensation cost is measured at fair value at date of grant and is expensed over the award’s vesting period with a corresponding increase in contributed surplus. For stock options granted to non-employees, the Corporation measures based on the fair value of services received, unless those are not reliably estimable, in which case the Corporation measures the fair value of the equity instruments granted. Compensation cost is measured when the Corporation obtains the goods or the counterparty renders the service.
| - 55 - |
Tax credits
Refundable tax credits related to eligible expenses are accounted for as a reduction of related costs in the year during which the expenses are incurred as long as there is reasonable assurance of their realization.
Financial Instruments
Credit Risk
Credit risk is the risk of a loss if a customer or counterparty to a financial asset fails to meet its contractual obligations. The Corporation has credit risk relating to cash, cash equivalents and short-term investments, which it manages by dealing only with highly-rated Canadian institutions. The carrying amount of financial assets, as disclosed in the statements of financial position, represents our credit exposure at the reporting date.
Currency risk
The Corporation is exposed to the financial risk related to the fluctuation of foreign exchange rates and the degrees of volatility of those rates. Foreign currency risk is limited to the portion of our business transactions denominated in currencies other than the Canadian dollar. Fluctuations related to foreign exchange rates could cause unforeseen fluctuations in our operating results.
A portion of the expenses, mainly related to research contracts and purchase of production equipment, is incurred in US dollars and in Euros, for which no financial hedging is required. There is a financial risk related to the fluctuation in the value of the US dollar and the Euro in relation to the Canadian dollar. In order to minimize the financial risk related to the fluctuation in the value of the US dollar in relation to the Canadian dollar, funds which were part of US dollar financings continue to be invested as short-term investments in the US dollar.
Furthermore, a portion of our cash and cash equivalents are denominated in US dollars, further exposing the Corporation to fluctuations in the value of the US dollar in relation to the Canadian dollar presented in Note 20 of the financial statements.
Interest rate risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market rates.
Our exposure to interest rate risk as at March 31, 2018, March 31, 2017, and February 28, 2017 is as follows:
| Cash and cash equivalents | Short-term fixed interest rate |
| Short-term investments | Short-term fixed interest rate |
| Unsecured convertible debentures | Long-term fixed interest rate |
The capacity of the Corporation to reinvest the short-term amounts with equivalent return will be impacted by variations in short-term fixed interest rates available on the market. Management believes the risk the Corporation will realize a loss as a result of the decline in the fair value of its short-term investments is limited because these investments have short-term maturities and are generally held to maturity.
Liquidity risk
Liquidity risk is the risk that the Corporation will not be able to meet its financial obligations as they fall due. The Corporation manages liquidity risk through the management of its capital structure and financial leverage, as outlined in Note 20 to the financial statements. It also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves our operating budgets, and reviews material transactions outside the normal course of business.
Our contractual obligations related to financial instruments and other obligations and liquidity resources are presented in the liquidity and capital resources of this MD&A.
Future Accounting changes
A number of new standards, interpretations and amendments to existing standards were issued by the International Accounting Standards Board (“IASB”) or the IFRS Interpretations Committee (IFRIC) that are mandatory but not yet effective for the period ended March 31, 2018 and have not been applied in preparing the financial statements. The following standards have been issued by the IASB with effective dates in the future that have been determined by management to impact the financial statements:
| - 56 - |
Financial instruments
On July 24, 2014, the International Accounting Standards Board (IASB) issued the final version of IFRS 9, Financial Instruments, replacing IAS 39, Financial Instruments: Recognition and Measurement. IFRS 9 introduces a revised approach for the classification of financial assets based on how an entity manages financial assets and the characteristics of the contractual cash flows of the financial assets replacing the multiple rules in IAS 39. Most of the requirements in IAS 39 for classification and measurement of financial liabilities have been carried forward in IFRS 9. IFRS 9 also introduces a new hedge accounting model that is more closely aligned with risk-management activities and a new expected credit loss model for calculating impairment on financial assets replacing the incurred loss model in IAS 39.
IFRS 9 is effective for annual periods beginning on or after January 1, 2018, with earlier adoption permitted. We intend to adopt IFRS 9 in its financial statements for the annual period beginning on April 1, 2018.
Our preliminary analysis has not identified any significant differences in respect to the classification and measurement of financial instruments and continues to evaluate the impact of the new standard on its financial statements.
Amendments to IFRS 2 – Classification and Measurement of Share-Based Payment Transactions
On June 20, 2016, the IASB issued amendments to IFRS 2, Share-Based Payment, clarifying how to account for certain types of share-based payment transactions. The amendments apply for annual periods beginning on or after January 1, 2018. Earlier application is permitted. As a practical simplification, the amendments can be applied prospectively. Retrospective, or early application is permitted if information is available without the use of hindsight. The amendments provide requirements on the accounting for: the effects of vesting and non-vesting conditions on the measurement of cash-settled share-based payments; share-based payment transactions with a net settlement feature for withholding tax obligations; and a modification to the terms and conditions of a share-based payment that changes the classification of the transaction from cash-settled to equity-settled. We intend to adopt the amendments to IFRS 2 in its financial statements for the annual period beginning on April 1, 2018. We have not yet assessed the impact of adoption of the amendments of IFRS 2.
| - 57 - |
| Item 6. | Directors, Senior Management and Employees |
| A. | Directors and Senior Management |
The following table sets out the name and the province or state and country of residence of each of our directors and all officers with us held by them, their principal occupation, the year in which they became a director, and the number of common shares they have declared to beneficially own, directly or indirectly, or over which control or direction is exercised by them.
| Name, Province or State, as the case may be, and Country or Residence of each Director | Principal Occupation | First Year as Director | Number of Common Shares Beneficially Owned or Controlled or Directed by Each Director(1) |
|
Roderick Carter California, United States Chairman of the Board |
Principal Aquila Life Sciences LLC | 2015 | - |
|
Jean-Marie (John) Canan Florida, United States |
Corporate Director | 2016 | 57,500 |
|
Janelle D’Alvise California, United States |
President and CEO of Acasti | 2016 | 52,500 |
|
Rick Schottenfeld(2) New York, New York |
Chairman of Schottenfeld Group Holding | 2017 | 50,000 |
|
Katherine Crewe(3) Quebec, Canada |
Managing Director, Canadian Operations, Mallinckrodt Pharmaceuticals | 2017 | - |
|
Donald Olds(4) Quebec, Canada |
President and CEO of NEOMED Institute | 2018 | - |
| _____________________________________________ | |
| Notes: | |
| (1) | Based on information publicly available on SEDI. |
| (2) | Mr. Schottenfeld resigned as a director on January 16, 2018. |
| (3) | Ms. Crewe resigned as a director on January 16, 2018. |
| (4) | Mr. Olds was appointed as a director on April 27, 2018. |
The following is a brief biography of our current directors and senior management:
Dr. Roderick N. Carter
Dr. Carter has a strong history of contributions to healthcare through clinical, research, business and people leadership. He has significant experience developing and commercializing nutraceutical and pharmaceutical products and has successfully led clinical research and business development strategies for cardiovascular and inflammation related diseases. Dr. Carter is currently Principal at Aquila Life Sciences LLC, a consulting firm he founded in April 2008 focusing on pharmaceutical development and commercialization. Prior to this, he was Vice President of Clinical Development at Reliant Pharmaceuticals, which developed the OM3 cardiovascular drug LOVAZA, and today is a wholly-owned subsidiary of GlaxoSmithKline. He also served as Executive Director at Merck and Co., USA, President and Chief Executive Officer of WellGen and Senior Medical Director at Pfizer Inc., USA. Dr. Carter received his Medical Degree from the University of Witwatersrand, Johannesburg, along with a Master of Science degree in Sports Medicine from Trinity College, Dublin.
Janelle D’Alvise (also our CEO)
Ms. D’Alvise has extensive experience in diagnostics, medical devices, pharmaceuticals and drug discovery research tools. Until recently, Ms. D’Alvise was the President and Chairman of Pediatric Bioscience. Before that, she was the CEO of Gish Biomedical, a cardiopulmonary medical device company. Prior to Gish, Ms. D’Alvise was the CEO of the Sidney Kimmel Cancer Center (SKCC), a drug discovery research institute. From 1995 until 1998, she was also the Co-Founder and Executive VP/COO of Metrika Inc., and in 1999 was the Co- Founder/President/CEO/Chairman of NuGEN, Inc. Ms. D’Alvise built both companies from technology concept through to successful regulatory approvals, product introduction and sustainable revenue growth. Prior to 1995, Ms. D’Alvise was a VP of Drug Development at Syntex/Roche and Business Unit Director of their Pain and Inflammation business, and also VP of Commercial Operations at SYVA, (Syntex’s clinical diagnostics division), and began her career with Diagnostic Products Corporation. Ms. D’Alvise has a B.S. in Biochemistry from Michigan Technological University. She has completed post- graduate work at the University of Michigan, Stanford University, and the Wharton Business Schools. Ms. D’Alvise has served on the board of numerous private companies and non-profits, and is an Entrepreneur-in-Residence for the von Liebig Institute for Entrepreneurship at the University of California, San Diego.
| - 58 - |
Jean-Marie (John) Canan
Mr. Canan is an accomplished business executive with over 34 years of strategic, business development and financial leadership experience. Mr. Canan recently retired from Merck & Co., Inc. where his last senior position was as Senior Vice-President, Global Controller, and Chief Accounting Officer for Merck from November 2009 to March 2014. He has managed all interactions with the audit committee of the Merck board of directors, while participating extensively with the main board and the compensation & benefits committee. Mr. Canan serves as a director of REV Group, a public company, where he chairs the audit committee. Mr. Canan also provides consulting services to Willow BioPharma, a Canadian start-up, engaged in the acquisition and development of legacy pharmaceutical assets. He also serves on the board of trustees of Angkor Hospital for Children, where he also chairs the audit & risk committee. Mr. Canan is a graduate of McGill University, Montreal, Canada, and is a Canadian Chartered Accountant.
Donald Olds
Mr. Olds is President and Chief Executive Officer of the NEOMED Institute, an R&D organization dedicated to advancing Canadian research discoveries to commercial success. Prior to NEOMED, he was the Chief Operating Officer of Telesta Therapeutics Inc., a TSX-listed biotechnology company, where he was responsible for finance and investor relations, manufacturing operations, business development, human resources and strategy. In 2016, he led the successful sale of Telesta to a larger public biotechnology company. Prior to Telesta, he was President and Chief Executive Officer of Presagia Corp., and Chief Financial Officer and Chief Operating Officer of Aegera Therapeutics, where he was responsible for clinical operations, business development, finance, and mergers and acquisitions. At both Telesta and Aegera, Mr. Olds was responsible for raising more than $100 million in equity financing and leading regional and global licensing transactions with life sciences companies. Mr. Olds is currently Director of Goodfood Market Corp, Chairman of Oxfam Quebec and Director of Presagia Corp. He has extensive past corporate governance experience serving on the boards of private and public for-profit and not-for-profit organizations. He holds an MBA (Finance & Strategy) and M.Sc. (Renewable Resources) from McGill University.
The following are brief biographies of our senior managers, other than our President and Chief Financial Officer, Janelle D’ Alvise, whose biography appears further above:
Linda P. O’Keefe – Chief Financial Officer (CFO)
Ms. O’Keefe has been our Chief Financial Officer since November 28, 2016. She has worked with both public and private biotechnology, diagnostics, medical devices and healthcare services firms, and also in other private equity-financed markets, including business services, education and technology. Prior to joining us, Ms. O’Keefe consulted with various firms after serving as Chief Financial Officer and executive-in-residence for Gryphon Investors, a San Francisco-based private equity firm. At Gryphon Investors, she led fundraising, limited partner relations, risk management and advised portfolio company management teams on growth, financing and back office strategies. In addition, Ms. O’Keefe provided mergers & acquisitions and integration support, established and led audit committees, and supported the expansion of teams and systems to meet the needs of growing companies. Ms. O’Keefe also served as Chief Financial Officer of Delphi Ventures, a healthcare-focused venture capital firm, and Elevate Ventures; as Vice President of Finance at Genelabs Technologies and Target Therapeutics; and as Controller at Collagen Corporation. Ms. O’Keefe is an active Certified Public Accountant and Chartered Global Management Accountant in California and Indiana and was formerly an audit senior with Ernst & Young. She is a member of the American Institute of CPAs, the California and Indiana Societies of CPAs, Association for Corporate Growth, Financial Executives International, and Healthcare Financial Management Association. Ms. O’Keefe holds a Bachelor of Science in Business from the University of California, Berkeley.
Dr. Pierre Lemieux – Chief Operating Officer (COO)
Dr. Lemieux has been our Chief Operating Officer since April 12, 2010. Previously, Mr. Lemieux was CEO, Co-Founder and Chairman of BiolActis Inc. which he sold in 2009 to interests affiliated with the Nestlé multinational group. Mr. Lemieux joined Suprateck Pharma in 1999 as Director and Vice-President involved in the development of formulations for gene therapy on behalf of Rhone-Poulenc Rorer and Genzyme, which today are under the Sanofi banner. Prior to this, Mr. Lemieux was involved in the development of cardiovascular products at Angiotech Pharmaceuticals. Mr. Lemieux has a Ph.D. in biochemistry from Université Laval (Québec). He holds 16 patents and has authored over 50 publications. Mr. Lemieux’s research was conducted at Université Laval as well as at the anti-cancer center Paul Papin D’Angers (France) and the University of Nottingham (England). His research focused on ovarian cancer and its treatment with monoclonal antibodies used to target cancer drugs. After completing his graduate studies, Mr. Lemieux joined the Oncology division of the Center for Health Research, University of Texas (U.S.). He obtained a postdoctoral fellowship from the Susan G. Komen Foundation (Breast Cancer). Mr. Lemieux has served on the boards of BioQuébec, Montreal in vivo and PharmaBio Development.
| - 59 - |
Mr. Brian Groch – Chief Commercial Officer
Mr. Groch has been our Chief Commercial Officer since June 4, 2018. Mr. Groch brings over 25 years of senior experience in the healthcare and life science industries, including product commercialization, developing and executing global sales strategies, business development, and operations. Most recently, Mr. Groch served as Executive Vice President and Chief Commercial Officer at Veru Inc., a urology, oncology and female health products company, where he was responsible for leading the development and execution of the company’s long-term commercial strategy. Under his leadership, Veru experienced rapid growth in sales of the company’s women’s health product. Mr. Groch also served as Chief Commercial Officer for Telesta Therapeutics, where he led the development and implementation of the global commercial strategy. Previously, Mr. Groch served as Vice President of Commercial Operations and Market Access for Horizon Therapeutics, where he oversaw global operations including the integration of two acquisitions valued over $1.5 billion. Mr. Groch has also served as CEO and President of Exsto Therapeutics, Head of Market Access for Dendreon, and Director of Health Policy for Phadia. He has held senior management roles with Novartis and Merck & Co. He holds an M.S. in Healthcare Administration and Marketing from Central Michigan University, as well as a B.S. in Physiology from Central Michigan University.
| B. | Compensation |
Summary of our Compensation Programs
Our executive compensation program is intended to attract, motivate and retain high-performing senior executives, encourage and reward superior performance and align the executives’ interests with ours by providing compensation which is competitive with the compensation received by executives employed by comparable companies and ensuring that the achievement of annual objectives is rewarded through the payment of bonuses and providing executives with long-term incentive through the grant of stock options.
Our governance & human resources, or GHR, committee has authority to retain the services of independent compensation consultants to advise its members on executive compensation and related matters, and to determine the fees and the terms and conditions of the engagement of those consultants. During our fiscal year ended March 31, 2018, the GHR committee retained compensation consulting services, including those led by The Sarkaria Group, to review our executive compensation programs, including base salary, short-term and long-term incentives, total cash compensation levels and total direct compensation of certain senior positions, against those of peer groups of similar and larger size, as measured by market capitalization, biotechnology and pharmaceutical companies listed or headquartered in North America. All of the services provided by the consultants were provided to the GHR committee. The GHR committee assessed the independence of the consultants and concluded that its engagement of the consultants did not raise any conflict of interest with us or any of our directors or executive officers. As influenced by the consultants’ fiscal period 2019 executive compensation review, the board and GHR committee set the following executive compensation program.
Use of Fixed and Variable Pay Components
Compensation of our named executive officers (“NEOs”) is revised each year and has been structured to encourage and reward executive officers on the basis of short-term and long-term corporate performance. In the context of its analysis of compensation for our fiscal year ended March 31, 2018, the following components were examined by the GHR committee:
| · | base salary; |
| · | short term incentive plan, consisting of a cash bonus; |
| · | long term incentive plan, consisting of stock options and equity incentive grants based on performance and/or time vesting conditions; and |
| · | other elements of compensation, consisting of group benefits and perquisites. |
Base Salary
We intend to be competitive with comparator companies and to attract and retain top talent. The GHR committee will review compensation periodically to be sure it meets this strategic imperative. Base salary is set to reflect an individual’s skills, experience and contributions within a salary structure consistent with our gender pay equity policy. Base salary structure is revised annually by the GHR committee as our financial and market conditions evolve.
| - 60 - |
Short Term Incentive Plan (STIP)
Our Short-Term Incentive Plan, or STIP, provides for potential rewards when a threshold of corporate performance is met. Personal objectives that support corporate goals are established annually with each employee and are assessed at the end of each financial year. Personal objectives are assessed through a performance grid, with pre-specified, objective performance criteria. STIP awards are paid out in proportion to individual performance, determined in end-of-year performance reviews. For the most senior participants in the STIP, greater weight is assigned to corporate objectives. Target payout is expressed as a percentage of base salary and is determined by employment contracts and board discretion. Annual salary for STIP purposes is the annual salary in effect at the end of the plan year (i.e., prior to annual salary increases).
The actual amount awarded ranges from zero for performance well below expectation and is capped at two times target for exceptional performance. The STIP is a discretionary variable compensation plan and all STIP payments are subject to board approval. Participants must be employed by us at the end of the financial year to qualify. We reserve the right to modify or discontinue the STIP at any time.
Ms. D’Alvise, our CEO, is eligible for up to a 50% bonus of her annual base salary and Ms. O’Keefe, our CFO, is eligible for up to a 40% bonus of her annual base salary. Dr. Lemieux, our COO, is eligible for up to a 40% bonus of his annual base salary and Mr. Groch, our Chief Commercial Officer, is eligible for up to a 40% bonus of his annual base salary.
These performance goals will take into account the achievement of R&D milestones within timelines and budget and individual objectives determined annually by the board according to short-term priorities.
Long Term Incentive Plan (LITP)
The LTIP has been adopted as a reward and retention mechanism. Participation is determined annually at the discretion of the board. Employees approved by our board of directors may participate in our stock option plan, which is designed to align the long-term interests of participants with those of shareholders, in order to promote shareholder value.
The GHR committee determines the number of stock options to be granted to a participant based on peer group data and taking into account corporate performance and level in the organization. The LTIP calculation is based on a guideline percentage of base salary and the number of options is determined based on an approved dollar value (rather than a specific number of shares). The guideline ranges from 15% to 200% and is subject to adjustment by the board in reviewing annual achievement of corporate performance and availability of shares. The GHR committee may also determine, in its sole discretion, ad hoc stock option awards to be granted to participants in order to address extraordinary situations. Awards at any level may be adjusted as necessary to maintain an equity burn rate and overhang similar to comparator companies. In addition to our stock option plan, the board is also empowered to grant ad hoc awards, from time to time, under our equity incentive plan to provide for a share-related mechanism to attract, retain and motivate qualified directors, senior employees and consultants.
Our directors and executive officers are not permitted to purchase financial instruments, such as prepaid variable forward contracts, equity swaps, collars or units of exchange funds that are designed to hedge or offset a decrease in market value of equity securities granted as compensation or held, directly or indirectly, by the director or officer.
Share Ownership Guidelines
To further align the interests of our executives with those of our other shareholders, the board has adopted share ownership guidelines. Under these guidelines, the CEO and other executives (i.e., CFO, COO, VPs) are required to retain and hold 50% of the shares acquired by them under any equity incentive award granted on or after June 8, 2017 (after subtracting shares sold to pay for option exercise costs, and relevant federal, state, and local taxes which are assumed to be at the highest marginal tax rates). In addition, the share retention rule applies unless the executive beneficially owns shares with a value at or in excess of the following share ownership guidelines:
| · | CEO — 2x then-current annual base salary |
| · | Other executives — 1x then-current annual base salary. |
The value of an individual’s shares for purposes of the share ownership guidelines is deemed to be the greater of the then- current fair market value of the shares, or the individual’s cost basis in the shares. Shares counted in calculating the share ownership guidelines include shares beneficially owned outright, whether from open market purchases, shares retained after option exercises, and shares of restricted stock or deferred stock units that have fully vested. In addition, in the case of vested, unexercised, in-the-money stock options, the in-the-money value of the stock options will be included in the share ownership calculation. Executives have five years from their date of hire or promotion to satisfy the share ownership guidelines.
| - 61 - |
Stock Option Plan
Our stock option plan was adopted by our board of directors on October 8, 2008 and has been amended from time to time, including most recently on June 14, 2017. The grant of options is part of the long-term incentive component of executive and director compensation and an essential part of compensation. Qualified directors, employees and consultants may participate in our stock option plan, which is designed to encourage option holders to link their interests with those of our shareholders, in order to promote an increase in shareholder value. Awards and the determination of any exercise price are made by our board of directors, after recommendation by the GHR committee. Awards are established, among other things, according to the role and responsibilities associated with the participant’s position and his or her influence over appreciation in shareholder value. Any award grants a participant the right to purchase a certain number of common shares during a specified term in the future, after a vesting period and/or specific performance conditions, at an exercise price equal to at least 100% of the market price (as defined below) of our common shares on the grant date. The “market price” of common shares as of a particular date generally means the closing price per common share on the TSXV, or any other exchange on which the common shares are listed from time to time, for the last preceding date on which there was a sale of common shares on that exchange (subject to certain exceptions set forth in the stock option plan in the event that we are no longer traded on any stock exchange). Previous awards may sometimes be taken into account when new awards are considered.
In accordance with the stock option plan, all of an option holder’s options will immediately vest on the date of a Change of Control event (as defined in the stock option plan), subject to the terms of any employment agreement or other contractual arrangement between the option holder and us.
However, in no case will the grant of options under the plan, together with any proposed or previously existing security based compensation arrangement, result in (in each case, as determined on the grant date): the grant to any one consultant within any 12-month period, of options reserving for issuance a number of common shares exceeding in the aggregate 2% of our issued and outstanding common shares (on a non-diluted basis); or the grant to any one employee, which provides investor relations services, within any 12-month period, of options reserving for issuance a number of common shares exceeding in the aggregate 2% of our issued and outstanding common shares (on a non-diluted basis).
Options granted under the stock option plan are non-transferable and are subject to a minimum vesting period of 18 months, with gradual and equal vesting on no less than a quarterly basis. They are exercisable, subject to vesting and/or performance conditions, at a price equal to the closing price of the common shares on the TSXV on the day prior to the grant of such options. In addition, and unless otherwise provided for in the agreement between us and the holder, options will also lapse upon termination of employment or the end of the business relationship with us except that they may be exercised for 60 days after termination or the end of the business relationship (30 days for investor relations services employees), to the extent that they will have vested on such date of termination of employment, except in the case of death, disability or retirement where this period is extended to 12 months.
Subject to the approval of relevant regulatory authorities, including the TSXV, if applicable, and compliance with any conditions attached to that approval (including, in certain circumstances, approval by disinterested shareholders) if applicable, the board of directors has the right to amend or terminate the stock option plan. However, unless option holders consent to the amendment or termination of the stock option plan in writing, any such amendment or termination of the stock option plan cannot affect the conditions of options that have already been granted and that have not been exercised under the stock option plan.
Options for common shares representing a fixed rate of 20% of our outstanding issued common shares as of February 29, 2016 may be granted by the board under the stock option plan. As at March 31, 2018, there were 2,940,511 common shares reserved for issuance under the stock option plan. As of March 31, 2018, there were 2,284,388 options outstanding under the stock option plan.
Equity Incentive Plan
On May 22, 2013, our equity incentive plan was adopted by the board in order to, among other things, provide us with a share- related mechanism to attract, retain and motivate qualified directors, employees and consultants. The adoption of the equity incentive plan was initially approved by shareholders at our 2013 Shareholders’ meeting held on June 27, 2013 and has been amended from time to time, including most recently on June 14, 2017.
Eligible persons may participate in the equity incentive plan. “Eligible persons” under the equity incentive plan consist of any director, officer, employee or consultant (as defined in the equity incentive plan) of us or a subsidiary. A participant is an eligible person to whom an award has been granted under the equity incentive plan. The equity incentive plan provides us with the option to grant to eligible persons bonus shares, restricted shares, restricted share units, performance share units, deferred share units and other share-based awards.
If, and for so long as our common shares are listed on the TSXV, no more than 2% of the issued and outstanding common shares may be granted to any one consultant or employee conducting investor relations activities in any 12-month period.
| - 62 - |
The board has the right to determine that any unvested or unearned restricted share units, deferred share units, performance share units or other share-based awards or restricted shares subject to a restricted period outstanding immediately prior to the occurrence of a change in control will become fully vested or earned or free of restriction upon the occurrence of a change in control. The board may also determine that any vested or earned restricted share units, deferred share units, performance share units or other share-based awards will be cashed out at the market price as of the date a change in control is deemed to have occurred, or as of such other date as the board may determine prior to the change in control. Further, the board has the right to provide for the conversion or exchange of any restricted share unit, deferred share unit, performance share unit or other share-based award into or for rights or other securities in any entity participating in or resulting from the change in control.
The equity incentive plan is administered by the board and the board has sole and complete authority, in its discretion, to determine the type of awards under the equity incentive plan relating to the issuance of common shares (including any combination of bonus shares, restricted share units, performance share units, deferred share units, restricted shares or other share-based awards) in such amounts, to such persons and under such terms and conditions as the board may determine, in accordance with the provisions of the equity incentive plan and the recommendations made by the GHR committee.
Subject to the adjustment provisions provided for in the equity incentive plan and the applicable rules and regulations of all regulatory authorities to which we are subject (including any stock exchange), the total number of common shares reserved for issuance pursuant to awards granted under the equity incentive plan will be equal to a number that (A) if, and for so long as the common shares are listed on the TSXV, will not exceed the lower of (i) 367,563 common shares, and (ii) 20% of the issued and outstanding common shares as of March 31, 2017, representing 2,940,511 common shares, which includes common shares issuable pursuant to options issued under our stock option plan.
Other Forms of Compensation
RRSP Matching Program. Effective June 1, 2016, we sponsor a voluntary Registered Retirement Savings Plan, or RRSP, matching program, which is open to all eligible employees, including NEOs. The RRSP matching program matches employees’ contributions up to a maximum of $1,500 per fiscal year for eligible employees who participate in the program. Other than matching contributions under the RRSP matching program (which amounts are disclosed in the column entitled “All Other Compensation” in the summary compensation table below), we do not provide pension or retirement benefits to our executive officers or directors.
Other Benefits and Perquisites. Our executive employee benefit program also includes life, medical, dental and disability insurance. These benefits and perquisites are designed to be competitive overall with equivalent positions in comparable organizations. We do not have a pension plan for employees.
| - 63 - |
Compensation Paid to Named Executive Officers
The following table sets forth the compensation information for the NEOs during the fiscal year ended March 31, 2018, the thirteen months ended March 31, 2017 and the fiscal year ended February 29, 2016.
| Name and Principal Position | Period ended |
Salary ($) |
Share-Based
Awards ($) |
Option-Based
Awards ($)(1) (2) |
Annual
Incentive Plans ($) |
All
Other Compensation ($) |
Total Compensation ($) |
|
Janelle
D’Alvise(4) President and CEO |
March 31, 2018 | 431,902 | - | 528,279 | 183,500(6) | - | 1,143,681 |
| March 31, 2017 | 365,072 | - | 502,163 | 136,049(7) | - | 1,003,284 | |
|
Linda
P. O’Keefe(5) CFO |
March 31, 2018 | 327,199 | - | 159,712 | 64,475(8) | - | 551,386 |
| March 31, 2017 | 114,183 | - | 237,340 | 39,897(9) | 109,414(10) | 500,834 | |
|
Pierre
Lemieux COO |
March 31, 2018 | 253,680 | - | 190,426 | 71,155 | 1,500(3) | 516,761 |
| March 31, 2017 | 275,819 | - | 96,522 | 49,000 | - | 421,341 | |
| February 29, 2016 | 239,565 | - | 33,320 | 42,000 | - | 314,885 | |
|
Laurent
Harvey VP, Clinical and Nonclinical Affairs |
March 31, 2018 | 187,642 | - | 135,141 | 46,698 | - | 369,481 |
| March 31, 2017 | 194,846 | - | 84,205 | 35,000 | - | 314,051 | |
| February 29, 2016 | 159,808 | - | 17,153 | 16,000 | - | 192,961 | |
____________________
Notes:
| (1) | The fair value of stock options is estimated at the grant date using the Black-Scholes option pricing model. This model requires the input of a number of parameters, including share price, share exercise price, expected share price volatility, expected time until exercise and risk-free interest rates. Although the assumptions used reflect management’s best estimates, they involve inherent uncertainties based on market conditions generally outside of our control. |
| (2) | The fair value of the option-based awards granted on June 14, 2017 in the fiscal year ended March 31, 2018 is $1.23. |
| (3) | The value of perquisites and other personal benefits received by these executives did not total an aggregate value of $50,000 or more, and does not represent 10% or more of their total salary during the fiscal years ended March 31, 2018, March 31, 2017 and February 29, 2016. |
| (4) | Ms. D’Alvise was appointed our President and CEO on May 11, 2016 and began her functions on June 1, 2016. Her employment agreement provides for payments in U.S. dollars with an annual base salary of US$330,250. In fiscal 2018, Ms. D’Alvise earned an annual base salary of US$338,250. |
| (5) | Ms. O’Keefe was appointed our CFO effective as of November 27, 2016. Her employment agreement provides for payments in U.S. dollars with an annual base salary of US$250,000. In fiscal 2018, Ms. O’Keefe earned an annual base salary of US$256,250. |
| (6) | US$142,303 converted as at March 31, 2018, based on a closing exchange rate of US$1.00 = $1.2895. |
| (7) | US$102,300, converted as at March 31, 2017, based on a closing exchange rate of US1.00= $1.3299. |
| (8) | US$50,000 converted as at March 31, 2018, based on a closing exchange rate of US$1.00 = $1.2895. Earned, but $US25,000 payable after FY 2019 event. |
| (9) | US$30,000 converted as at March 31, 2017, based on a closing exchange rate of US1.00= $1.3299. |
| (10) | Consulting services from July 2016 to November 2016 which provided for payments in U.S. dollars: US$82,273, converted as at March 31, 2017 based on a closing exchange rate of US1.00= $1.3299. |
| - 64 - |
Outstanding Share-Based and Option-Based Awards
The following tables provide information about the number and value of the outstanding option-based awards held by the NEOs as of March 31, 2018. There are no share-based awards outstanding as of the date of this annual report.
| Name/Grant Date |
Number of Securities Underlying Unexercised Options (#) |
Option Exercise Price ($)(1) |
Option Expiration Date |
Value of Unexercised In-The-Money Options ($)(2) |
| Janelle D’Alvise(3) | ||||
| June 14, 2017 | 430,000 | 1.77 | June 14, 2027 | -- |
| May 12, 2016 | 525,000 | 1.56 | May 12, 2023 | -- |
| Linda P. O’Keefe(4) | ||||
| June 14, 2017 | 130,000 | 1.77 | June 14, 2027 | -- |
| February 24, 2017 | 200,000 | 1.65 | February 24, 2027 | -- |
| Pierre Lemieux | ||||
| June 14, 2017 | 155,000 | 1.77 | June 14, 2027 | -- |
| February 24, 2017 | 50,000 | 1.65 | February 24, 2027 | -- |
| May 30, 2016 | 31,400 | 1.99 | May 29, 2023 | -- |
| June 1, 2015 | 16,900 | 4.50 | June 1, 2022 | -- |
| October 20, 2014 | 7,500 | 6.50 | October 19, 2019 | -- |
| Laurent Harvey | ||||
| June 14, 2017 | 110,000 | 1.77 | June 14, 2027 | -- |
| February 24, 2017 | 50,000 | 1.65 | February 24, 2027 | -- |
| May 30, 2016 | 21,000 | 1.99 | May 29, 2023 | -- |
| June 1, 2015 | 8,700 | 4.50 | June 1, 2022 | -- |
| October 20, 2014 | 2,500 | 6.50 | October 19, 2019 | -- |
____________________
Notes:
| (1) | Option-based awards were consolidated following our share consolidation. The exercise price was increased proportionally to reflect the consolidation. | |
| (2) | Calculation is based on a trading price of $1.30 of our common shares on the TSXV, as at closing on March 29, 2018. | |
| (3) | Ms. D’Alvise was appointed as our President and CEO on May 11, 2016 and began her functions on June 1, 2016. | |
| (4) | Ms. O’Keefe was appointed as our CFO effective November 27, 2016. |
| - 65 - |
The following table sets out the value of share-based, option-based, and warrant-based awards held by the NEOs that vested during the fiscal year ended March 31, 2018:
| Name |
Share-Based Awards ($) |
Option-Based Awards ($) |
| Janelle D’Alvise | -- | 154,323 |
| Linda P. O’Keefe | -- | 74,787 |
| Pierre Lemieux | -- | 42,602 |
| Laurent Harvey | -- | 32,805 |
Compensation of Directors
Our directors’ compensation consists of an annual fixed compensation of US$30,000. While our compensation structure does not include meeting fees, a discretionary reduction of 20% may be applied to the annual retainer payment each time a director fails to attend a quarterly board or committee session. In addition, the chairman of the board and the chairman of the audit committee received additional compensation of US$30,000 and US$15,000, respectively, for their additional work during the fiscal year ended March 31, 2018. The directors are also entitled to be reimbursed for travelling and other reasonable expenses properly incurred by them in attending meetings of the board or any committee or in otherwise serving us, in accordance with our policy on travel and expenses.
Following their first election to our board of directors, non-executive directors are eligible to receive an initial equity grant of up to 150% of their annual cash retainer worth of stock options vesting annually in equal installments over a 3-year period, subject to the other terms and conditions set forth under the heading “Stock Option Plan”. In addition to their initial grant, non-executive directors are eligible to receive an annual equity-based award equal to 100% of their total annual cash retainer vesting quarterly in equal installments over an 18-month period. These awards will be granted at the same time that we are performing our annual performance review for our employees, subject to availability of common shares and subject to the terms and conditions described under the headings “Stock Purchase Plan” and “Equity Incentive Plan”. The level of these awards will be consistent with equivalent awards in comparable companies obtained from the benchmark exercise and in accordance with the recommendations obtained from our independent compensation consultant. The total compensation for our non-executive directors during fiscal year ended March 31, 2018 was as follows:
| Name | Fiscal Year Ended March 31, |
Fees Earned ($) |
Option-Based Awards ($)(1)(2) |
All Other Compensation ($)(3) |
Total ($) |
| Roderick N. Carter | 2018 | 75,627 (4) | 62,656 | -- | 138,283 |
| Jean-Marie (John) Canan | 2018 | 56,720 (5) | 35,628 | -- | 92,348 |
| Rick Schottenfeld | 2018 | 18,315 (6) | 55,124 | -- | 18,315 |
| Katherine Crewe | 2018 | 18,315 (6) | 55,124 | -- | 18,315 |
| Leendert Staal | 2018 | 21,315 (7) | 35,628 | -- | 21,315 |
____________________
Notes:
| (1) | The fair value of the awards is estimated at the grant date using the Black-Scholes option pricing model. This model requires the input of a number of parameters, including share price, share exercise price, expected share price volatility, expected time until exercise and risk-free interest rates. Although the assumptions used reflect management’s best estimates, they involve inherent uncertainties based on market conditions generally outside of our control. |
| (2) | For the fiscal year ended on March 31, 2018, the fair market value of the June 14, 2017 option-based awards is based on a fair value of $1.23 per option granted to Dr. Carter, Mr. Canan and Dr. Staal and the fair market value of $1.10 for option-based awards granted to Mr. Schottenfeld and Ms. Crewe on August 31, 2017. |
| (3) | The directors do not receive pension benefits or other non-equity based annual compensation. |
| (4) | Dr. Carter earned a director compensation of US$60,000. |
| (5) | Mr. Canan earned a director compensation of US$45,000. |
| (6) | Ms. Crewe and Mr. Schottenfeld earned a director compensation of US$14,583 from August 15, 2017 to January 15, 2018. |
| (7) | Dr. Staal earned a director compensation of US$16,875 to August 15, 2017. |
| - 66 - |
The following table provides information about the number and value of the outstanding share-based and option-based awards held by non-executive directors. There were no share-based awards outstanding as of the date of this annual report.
| Name/Grant Date |
Number of Securities Underlying Unexercised Options (#) |
Option Exercise Price ($)(1) |
Option Expiration Date |
Value of Unexercised In-The-Money Options ($)(2) |
| Roderick N. Carter | ||||
| June 14, 2017 | 51,000 | 1.77 | June 14, 2027 | -- |
| May 30, 2016 | 200,000 | 1.99 | May 29, 2023 | -- |
| August 19, 2015 | 10,000 | 4.80 | August 19, 2022 | -- |
| Jean-Marie (John) Canan | ||||
| June 14, 2017 | 29,000 | 1.77 | June 14, 2027 | -- |
| February 24, 2017 | 50,000 | 1.65 | February 24, 2027 | -- |
____________________
Notes:
| (1) | Option-based awards were consolidated following our share consolidation. The exercise price was increased proportionally to reflect the consolidation. | |
| (2) | Calculation is based on a trading price of $1.30 for our common shares on the TSXV, as at closing on March 29, 2018. |
None of the share-based and stock options of the Corporation held by non-executive Directors that vested during the financial year ended on March 31, 2018 were in-the-money at their respective vesting date.
| C. | Board Practices |
Board of Directors
Director Independence
Our board of directors believes that, in order to maximize its effectiveness, the board must be able to operate independently. A majority of directors must satisfy the applicable tests of independence, such that the board of directors complies with all independence requirements under applicable corporate and securities laws and stock exchange requirements applicable to us. No director will be independent unless the board of directors has affirmatively determined that the director has no material relationship with us or any of our affiliates, either directly or indirectly or as a partner, shareholder or officer of an organization that has a relationship with us or our affiliates. Such determinations will be made on an annual basis and, if a director joins the board of directors between annual meetings, at such time.
Independent Directors
The board of directors determined that Mr. Canan, Dr. Carter and Mr. Olds are independent within the meaning of NI 52-110 and NASDAQ Stock Market rules.
Directors Who are Not Independent
The board of directors determined that Ms. D’Alvise is not independent within the meaning of NI 52-110 and NASDAQ given that she is our President and CEO.
During the fiscal year ended March 31, 2018, the board of directors held 13 meetings. Attendance of directors at those meetings is indicated in the table below:
| Board Members(1) | Attendance |
| Roderick N. Carter | 13 out of 13 |
| Jean-Marie (John) Canan | 13 out of 13 |
| Janelle D’Alvise | 13 out of 13 |
____________________
Note:
| (1) | This table excludes directors who resigned from the board during the course of the fiscal year ended March 31, 2018. |
Chairman of the Board
Dr. Carter acts as Chairman of the board. His duties and responsibilities consist of the oversight of the quality and integrity of the board of directors’ practices.
| - 67 - |
Board Mandate
There is no specific mandate for the board of directors, since the board has plenary power. Any responsibility that is not delegated to senior management or a committee of the board remains with the full board of directors.
Position Descriptions
No written position description has been approved for the chair of the board of directors and for the chairs of each committee. The primary role and responsibility of the chair of each committee of the board of directors is to: (i) in general, ensure that the committee fulfills its mandate, as determined by the board of directors; (ii) chair meetings of the committee; (iii) report to the board of directors; and (iv) act as liaison between the committee and the board of directors and, if necessary, our management.
Orientation and Continuing Education
We provide orientation for new appointees to the board of directors and committees in the form of informal meetings with members of the board and senior management, complemented by presentations on the main areas of our business. The board does not formally provide continuing education to its directors, as directors are experienced members. The board of directors relies on professional assistance, when judged necessary, in order to be educated/updated on a particular topic.
Code of Business Conduct and Ethics
The board of directors adopted a Code of Business Conduct and Ethics, or Code of Conduct, for our directors, officers and employees on May 31, 2007, as amended from time to time. Our Code of Conduct can be found on SEDAR at www.sedar.com and on our web site on www.acastipharma.com. A copy of the Code of Conduct can also be obtained by contacting our Corporate Secretary. Since its adoption by the board of directors, any breach of the Code of Conduct must be brought to the attention of the board of directors by our CEO or other senior executives. No report has ever been filed which pertains to any conduct of a director or executive officer that constitutes a breach to our Code of Conduct.
Since the adoption of the Code of Conduct and the following policies, the board of directors actively monitors compliance with the Code Conduct and promotes a business environment where employees are encouraged to report malfeasance, irregularities and other concerns. The Code of Conduct provides for specific procedures for reporting non-compliant practices in a manner which, in the opinion of the board of directors, encourages and promotes a culture of ethical business conduct.
The board of directors also adopted a disclosure policy, insider trading policy, majority voting policy, management and board compensation policies, and a whistleblower policy.
In addition, under the Civil Code of Québec, to which we are subject as a legal person incorporated under the Business Corporations Act (Québec) (L.R.Q., c. S-31), a director o must immediately disclose to the board any situation that may place him or her in a conflict of interest. Any such declaration of interest is recorded in the minutes of proceeding of the board of directors. The director abstains, except if required, from the discussion and voting on the question. In addition, it is our policy that an interested director recuse himself or herself from the decision-making process pertaining to a contract or transaction in which he or she has an interest.
Nomination of Directors
The board of directors receives recommendations from the GHR committee, but retains responsibility for managing its own affairs by, among other things, giving its approval for the composition and size of the board of directors, and the selection of candidates nominated for election to the board of directors. The GHR committee initially evaluates candidates for nomination for election as directors, having regard to the background, employment and qualifications of possible candidates.
The selection of the nominees for the board of directors is made by the other members of the board, based on our needs and the qualities required for the board of directors, including ethical character, integrity and maturity of judgment of the candidates; the level of experience of the candidates, their ideas regarding the material aspects of our business, the expertise of the candidates in fields relevant to us while complementing the training and experience of the other members of the board of directors; the will and ability of the candidates to devote the necessary time to their duties to the board of directors and its committees, the will of the candidates to serve on the board of directors for numerous consecutive financial periods and finally, the will of the candidates to refrain from engaging in activities which conflict with the responsibilities and duties of a director. The board researches the training and qualifications of potential new directors which seem to correspond to the selection criteria of the board of directors and, depending on the results of said research, organizes meetings with the potential candidates.
In the case of incumbent directors whose terms of office are set to expire, the board will review such directors’ overall service to us during their term of office, including the number of meetings attended, level of participation, quality of performance and any transactions of such directors with us during their term of office.
| - 68 - |
We may use various sources in order to identify the candidates for the board of directors, including our own contacts and the references of other directors, officers, advisors and executive placement agencies. We will consider director candidates recommended by shareholders and will evaluate those director candidates in the same manner in which we evaluate candidates recommended by other sources. In making recommendations for director nominees for the annual meeting of shareholders, we will consider any written recommendations of director candidates by shareholders received by our Corporate Secretary not later than 120 days before the anniversary of the previous year’s annual meeting of shareholders. Recommendations must include the candidate’s name, contact information and a statement of the candidate’s background and qualifications, and must be mailed to us. Following the selection of the candidates by the board of directors, we will propose a list of candidates to the shareholders, for our annual meeting of shareholders.
The board of directors does not have a nominating committee and has not adopted any formal written director term limit policy. Proposed nominations of director candidates are evaluated by our GHR committee.
GHR Committee
The mandate of the GHR committee consists of the evaluation of the proposed nominations of senior executives and director candidates to our board of directors, recommending for board approval, if appropriate, revisions of our corporate governance practices and procedures, developing new charters for any new committees established by the board of directors, monitoring relationships and communication between management and the board of directors, monitoring emerging best practices in corporate governance and oversight of governance matters and assessing the board of directors and its committees. The GHR committee is also in charge of establishing the procedure which must be followed by us to comply with applicable guidelines of the TSXV and NASDAQ Stock Market regarding corporate governance.
The GHR committee has the responsibility of evaluating the compensation, performance incentives as well as the benefits granted to our upper management in accordance with their responsibilities and performance as well as to recommend the necessary adjustments to our board of directors. The GHR committee also reviews the amount and method of compensation granted to the directors. The GHR committee may retain an external firm in order to assist it during the execution of its mandate. The GHR committee considers time commitment, comparative fees and responsibilities in determining compensation.
The GHR committee is composed of independent members within the meaning of NI 52-110 and NASDAQ Stock Exchange rules, namely Dr. Carter, Mr. Canan and Mr. Olds.
Periodic Assessments
The board of directors, its committees and each director are subject to periodic evaluations of their efficacy and contribution. The evaluation procedure consists in identifying any shortcomings and implementing adjustments proposed by directors at the beginning and during meetings of the board of directors and of each of its committees. Among other things, these adjustments deal with the level of preparation of directors, management and consultants employed by us, the relevance and sufficiency of the documentation provided to directors and the time allowed to directors for discussion and debate of items on the agenda.
Director Term Limits
The board actively considers the issue of term limits from time to time. At this time, the board does not believe that it is in our best interests to establish a limit on the number of times a director may stand for election. While such a limit could help create an environment where fresh ideas and viewpoints are available to the board, a director term limit could also disadvantage us through the loss of the beneficial contribution of directors who have developed increasing knowledge of, and insight into, us and our operations over a period of time. As we operate in a unique industry, it is difficult to find qualified directors with the appropriate background and experience and the introduction of a director term limit would impose further difficulty.
Policies Regarding the Representation of Women on the Board and Among Executive Officers
We have not adopted a formal written policy regarding diversity amongst executive officers and members of the board of directors, including mechanisms for board renewal, in connection with, among other things, the identification and nomination of women directors. Nevertheless, we recognize that gender diversity is a significant aspect of diversity and acknowledges the important role that women with appropriate and relevant skills and experience can play in contributing to the diversity of perspective on the board of directors.
Rather than considering the level of representation of women for directorship and executive officer positions when making board or executive officer appointments, we consider all candidates based on their merit and qualifications relevant to the specific role. While we recognize the benefits of diversity at all levels within its organization, we do not currently have any targets, rules or formal policies that specifically require the identification, consideration, nomination or appointment of candidates for directorship or executive management positions or that would otherwise force the composition of our board of directors and executive management team. Currently, we have one women director who is also our CEO. In addition, our CFO is a woman.
| - 69 - |
Audit Committee
Our audit committee is responsible for assisting the board of directors in fulfilling its oversight responsibilities with respect to financial reporting, including:
| · | reviewing our procedures for internal control management performing financial functions; |
| · | reviewing and approving the engagement of the auditor; |
| · | reviewing annual and quarterly financial statements and all other material continuous disclosure documents, including our annual information form and management’s discussion and analysis; |
| · | assessing our financial and accounting personnel; |
| · | assessing our accounting policies; |
| · | reviewing our risk management procedures; and |
| · | reviewing any significant transactions outside our ordinary course of business and any pending litigation involving us. |
The audit committee has direct communication channels with our management performing financial functions and our external auditor to discuss and review such issues as the audit committee may deem appropriate. As of March 31, 2018, the audit committee was composed of Mr. Canan, as chairperson Dr. Carter and Ms. D’Alvise. Each of Mr. Canan and Dr. Carter is “financially literate” and “independent” within the meaning of NI 52-110 and the Exchange Act. In accordance with the exemption provided under Section 3.5 of NI 52-110, Ms. D’Alvise acted as a member of the audit committee from January 16, 2018 to April 27, 2018 in order to fill a vacancy resulting from the resignation of Mr. Schottenfeld on January 16, 2018. As of the date of this annual report, the audit committee is composed of independent members within the meaning of NI 52-110 and the Exchange Act, namely Mr. Canan, Dr. Carter and Mr. Olds.
Compensation Governance
Compensation of our executive officers and directors is recommended to the board of directors by the GHR committee. In its review process, the GHR committee relies on input from management on the assessment of executives and corporate performance. During the fiscal year ended March 31, 2018, the GHR committee was composed of the following members, each of whom is independent: Dr. Carter and Mr. Canan. The GHR committee establishes management compensation policies and oversees their general implementation. All members of the GHR committee have direct experience which is relevant to their responsibilities as GHR committee members. All members are or have held senior executive or director roles within significant businesses, several also having public companies experience, and have a good financial understanding which allows them to assess the costs versus benefits of compensation plans. The members combined experience in our sector provides them with the understanding of our success factors and risks, which is very important when determining metrics for measuring success.
Risk management is a primary consideration of the GHR committee when implementing its compensation program. We do not believe that our compensation program results in unnecessary or inappropriate risk taking, including risks that are likely to have a material adverse effect on us. Payments of bonuses, if any, are not made unless performance goals are met.
For executives, more than half of target direct compensation (base salary + target STIP awards + target LTIP awards) is considered “at risk”. We believe this mix results in a strong pay-for-performance relationship and an alignment with shareholders and is competitive with other firms of comparable size in similar fields. The CEO (or any person acting in that capacity) makes recommendations to the GHR committee as to the compensation of our executive officers, other than himself or herself, for approval by the board. The GHR committee makes recommendations to the board of directors as to the compensation of the CEO, for approval. The CEO’s salary is based on comparable market consideration and the GHR committee’s assessment of his or her performance, with regard to our financial performance and progress in achieving strategic goals.
Qualitative factors beyond the quantitative financial metrics are also a key consideration in determination of individual executive compensation payments. How executives achieve their financial results and demonstrate leadership consistent with our values are key to individual compensation decisions.
| D. | Employees |
Our management consists of professionals experienced in business development, finance and science. Our research team includes scientists with expertise in pharmaceutical development, chemistry, manufacturing and controls, nonclinical and clinical studies, pharmacology, regulatory affairs, quality assurance/quality control, intellectual property and strategic alliances. As of March 31, 2018, we had 20 full-time employees. We generally require all of our employees to enter into an invention assignment, non-disclosure and non-compete agreement. We rely, in part, on the administrative and other staff of Neptune and also rely on consultants from time to time. Our employees are not covered by any collective bargaining agreement or represented by a trade union. We consider our relations with our employees to be good and our operations have never been interrupted as the result of a labor dispute.
| - 70 - |
| E. | Share Ownership |
The following table shows the total number of common shares beneficially owned by each of our directors and executive officers and the percentage of the total issued and outstanding common shares that such holdings represent.
| Common shares beneficially owned | Percentage of total issued and outstanding common shares |
|||||||
| Name | as of March 31, 2018 | as of March 31, 2018(1) | ||||||
| Roderick N. Carter | - | - | ||||||
| Jean-Marie (John) Canan | 57,500 | * | ||||||
| Janelle D’Alvise | 52,500 | * | ||||||
| Linda P. O’Keefe | 30,000 | * | ||||||
| Pierre Lemieux | 7,000 | * | ||||||
| Laurent Harvey | - | - | ||||||
| Donald Olds | - | - | ||||||
(1) Based on 36,628,063 common shares outstanding as of the date of this annual report.
* Less than 1%.
See “Item 6.B. Compensation” above for information regarding the share-based, option-based, call-option-based, and warrant- based awards held by our directors and executive officers and for a description of our stock option plan and equity incentive plan.
| Item 7. | Major Shareholders and Related Party Transactions |
| A. | Major Shareholders |
As of the date of this annual report, and based on information publicly available to us on SEDI (www.sedi.ca), Neptune owns 5,064,694 common shares representing 13.8% of our common shares issued and outstanding. The common shares are voting, participating, and have no par value. Neptune also owns warrants entitling it to acquire 592,500 common shares (in order to obtain 1 common share, 10 warrants must be exercised). Neptune does not have different voting rights than other holders of common shares. To the best of our knowledge, there are no other beneficial owners of 5% or more of any class of our voting securities.
All common shares, including those held by Neptune, are common shares with the same voting rights. Based on the records of our registrar and transfer agent, Computershare Investor Services Inc., as of the date of this annual report, there are approximately 8 registered holders (including The Depository Trust Company) of our common shares resident in the United States (approximately 32% of all registered holders).
| B. | Related Party Transactions |
Please see the section entitled “—Related Party Transactions” in “Item 5. Operating and Financial Review and Prospects”.
| C. | Interests of Experts and Counsel |
Not applicable.
| Item 8. | Financial Statements |
| A. | Financial Statements and Other Financial Information |
Financial Statements
See “Item 17. Financial Statements” for our audited financial statements.
Legal Proceedings
Due to the fact that a significant portion of our intellectual property rights are licensed to us by Neptune, we rely on Neptune to protect a significant portion of the intellectual property rights that we use under our license agreement with Neptune. Neptune is engaged in a number of legal actions related to its intellectual property.
| - 71 - |
Our former CEO is claiming the payment of approximately $8.5 million and the issuance of equity instruments from the Neptune group. As our management believes that these claims are not valid, no provision has been recognized. Neptune and its subsidiaries also filed an additional claim to recover certain amounts from the former officer.
We are also involved in other matters arising in the ordinary course of our business. Since management believes that all related claims are not valid and it is presently not possible to determine the outcome of these matters, no provisions have been made in our financial statements for their ultimate resolution beyond the amounts incurred and recorded for such matters. The resolution of these other matters could have an effect on our financial statements in the year that a determination is made, however, in management’s opinion, the final resolution of all such matters is not projected to have a material adverse effect on our financial position.
Dividend Policy
We do not anticipate paying any cash dividend on the common shares in the foreseeable future. We presently intend to retain future earnings to finance the expansion and growth of our business. Any future determination to pay dividends will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements and other factors the board of directors deems relevant. In addition, the terms of any future debt or credit facility may preclude us from paying dividends.
| Item 9. | The Offer and Listing |
| A. | Listing Details |
Since March 31, 2011, our common shares have been listed on the TSX-V under the ticker symbol ACST. Since January 7, 2013, our common shares have been listed on the NASDAQ Stock Market under the ticker symbol ACST. The following tables set forth, for the periods indicated, the high and low market prices of our common shares as reported on the TSX-V and the NASDAQ Stock Market.
(a) For the five most recent full fiscal years:
| Fiscal Year Ended | TSX-V | NASDAQ Stock Market | ||||||||||||||
| High ($) | Low ($) | High (US$) | Low (US$) | |||||||||||||
| February 28, 2014(1) | 43.20 | 16.00 | 39.90 | 20.00 | ||||||||||||
| February 28, 2015(1) | 14.90 | 11.50 | 13.40 | 10.90 | ||||||||||||
| February 29, 2016 | 7.60 | 1.83 | 6.10 | 1.30 | ||||||||||||
| March 31, 2017 | 4.03 | 1.47 | 3.09 | 1.11 | ||||||||||||
| March 31, 2018 | 2.75 | 1.16 | 3.10 | 0.9287 | ||||||||||||
________________________
Note:
| (1) | Our common shares were consolidated on October 15, 2015, on the basis of one (1) post-consolidation common share for every 10 pre-consolidation common shares, and each fractional common share resulting from the consolidation was rounded up. The common share price was increased proportionally to reflect the consolidation. |
| (b) | For each full financial quarter of the two most recent full fiscal years and any subsequent period: |
| Period | TSX-V | NASDAQ Stock Market | ||||||||||||||
| High ($) | Low ($) | High (US$) | Low (US$) | |||||||||||||
| 1st Quarter ended May 31, 2016 | 2.45 | 1.50 | 1.88 | 1.20 | ||||||||||||
| 2nd Quarter ended August 31, 2016 | 2.25 | 1.66 | 1.79 | 1.21 | ||||||||||||
| 3rd Quarter ended November 30, 2016 | 4.03 | 1.62 | 3.09 | 1.20 | ||||||||||||
| Four-month period ended March 31, 2017 | 2.66 | 1.47 | 2.03 | 1.11 | ||||||||||||
| 1st Quarter ended June 30, 2017 | 1.90 | 1.66 | 1.47 | 1.23 | ||||||||||||
| 2nd Quarter ended September 30, 2017 | 1.77 | 1.57 | 1.42 | 1.26 | ||||||||||||
| 3rd Quarter ended December 31, 2017 | 2.75 | 1.20 | 3.10 | 0.96 | ||||||||||||
| 4th Quarter ended March 31, 2018 | 1.40 | 1.16 | 1.26 | 0.9287 | ||||||||||||
| - 72 - |
| (c) | For the most recent six months: |
| Period | TSX-V | NASDAQ Stock Market | ||||||||||||||
| High ($) | Low ($) | High (US$) | Low (US$) | |||||||||||||
| November 2017 | 2.75 | 1.58 | 3.10 | 1.24 | ||||||||||||
| December 2017 | 2.14 | 1.20 | 1.68 | 0.9428 | ||||||||||||
| January 2018 | 1.57 | 1.16 | 1.26 | 0.9287 | ||||||||||||
| February 2018 | 1.36 | 1.24 | 1.09 | 0.97 | ||||||||||||
| March 2018 | 1.40 | 1.20 | 1.07 | 0.946 | ||||||||||||
| April 2018 | 1.48 | 0.98 | 1.13 | 0.77 | ||||||||||||
| May 2018 | 1.07 | 0.81 | 0.8303 | 0.63 | ||||||||||||
The holders of common shares are entitled to vote at all meetings of our shareholders except meetings at which only holders of a specified class or series of shares are entitled to vote. The holders of common shares are entitled to receive dividends as and when declared by the board, if any.
No common shares have been issued subject to call or assessment. There are no pre-emptive or conversion rights and no provisions for redemption or purchase for cancellation, surrender, or sinking or purchase funds. Our common shares must be issued as fully-paid and non-assessable, and are not subject to further capital calls by us. All of the common shares rank equally as to voting rights, participation in a distribution of our assets on a liquidation, dissolution or winding-up, and the entitlement to dividends. Common shares are transferable at the offices of our transfer agent and registrar, Computershare Investor Services Inc., in Toronto, Ontario, Canada and Montreal, Québec, Canada. There are no restrictions in our corporate documents on the free transferability of the common shares.
| B. | Plan of Distribution |
Not applicable.
| C. | Markets |
Since March 31, 2011, the common shares have been listed on the TSX-V under the ticker symbol ACST. Since January 7, 2013, the common shares have been listed on the NASDAQ Stock Market under the ticker symbol ACST.
| D. | Selling Shareholders |
Not applicable.
| E. | Dilution |
Not applicable.
| F. | Expenses of the Issuer |
Not applicable.
| Item 10. | Additional Information |
| A. | Share Capital |
Not applicable.
| - 73 - |
| B. | Memorandum and Articles of Association |
We were incorporated on February 1, 2002 under Part 1A of the Companies Act (Québec) under the name “9113-0310 Québec Inc”. On August 7, 2008, pursuant to a Certificate of Amendment, we changed our name to “Acasti Pharma Inc.”, our share capital, the provisions regarding the restrictions on securities transfers and our borrowing powers. On November 7, 2008, pursuant to a Certificate of Amendment, we further revised our provisions regarding our borrowing powers. We became a reporting issuer in Québec on November 17, 2008. On February 14, 2011, the Business Corporations Act (Québec) came into effect and replaced the Companies Act (Québec). We are now governed by the Business Corporations Act (Québec), or the BCA.
Register, Entry Number and Purposes
Our articles of incorporation, as amended, or Articles, and general by-laws, do not define any of our objects and purposes. In that respect, we have no limit on the type of business we can carry out.
Directors’ Powers
Our Articles and by-laws do not contain any provision regarding: (a) a director’s power in the absence of an independent quorum, to vote compensation to itself or any members of the committees of the board; (b) retirement or non-retirement of directors under an age limit requirement; and (c) number of shares, if any, required for a director’s qualification.
Our by-laws provide that a director may not vote on a resolution to approve, amend or terminate a contract or transaction in which the director has any financial stake that may reasonably be considered to influence decision-making or be present during deliberations concerning the approval, amendment or termination of such a contract or transaction, unless the contract or transaction: (a) relates primarily to the remuneration of the director or an associate of the director as a director of us or an affiliate of us, (b) relates primarily to the remuneration of the director or an associate of the director as an officer, employee or mandatary of us or an affiliate of us, if we are not a reporting issuer, (c) is for indemnity or liability insurance, or (d) is with an affiliate of us, and the sole interest of the director is as a director or officer of the affiliate. In addition, our by-laws provide that a director must avoid placing himself or herself in any situation where his or her personal interests would be in conflict with his obligations as a director of ours, and that a director must disclose to us any interest he or she has in a business or association that may place him or her in a situation of conflict of interest and of any right he or she may set up against us, indicating their nature and value, where applicable.
Our Articles provide that the board may, on behalf us, (a) borrow money, (b) issue, reissue, sell or pledge debt instruments, (c) guarantee the obligations of a third party, and (d) hypothecate all or any of its assets, both present and future, to guarantee the performance of any of our obligations.
The quorum at every meeting of the board has been set to the minimum number of directors required under our Articles. In the absence of a quorum, a director has no power to make any decision regarding, among other things, compensation to himself or herself or to any member of the committees of the board.
Our by-laws do not contain any requirements with respect to a mandatory retirement age for our directors and the number of shares required for directors’ qualifications.
Rights, Preferences and Restrictions Attaching to Each Class of Shares
Our authorized capital consists of an unlimited number of no par value common shares and an unlimited number of no par value Class B, Class C, Class D and Class E preferred shares (collectively, the preferred shares), issuable in one or more series. As of March 31, 2018, there were (i) a total of 25,638,215 common shares issued and outstanding and no preferred shares issued and outstanding, (ii) 2,284,388 options to purchase common shares issued and outstanding, at a weighted average exercise price of $1.81 per common share, (iii) 18,400,000 Series 8 public offering warrants issued in connection with our 2014 public offering at an exercise price of US$15.00 per common share (10 Series 8 public offering warrants must be exercised in order to acquire one common share), (iv) 161,654 Series 9 private placement warrants issued in connection with our 2014 private placement at an exercise price of $13.30 per common share, (v) $2,000,000 aggregate principal amount of unsecured convertible debentures, maturing on February 21, 2020, issued in our February 2017 private placement and contingent warrants to acquire 1,052,630 common shares (the debentures are convertible into up to 1,052,630 common shares at any time by the holders at a fixed price of $1.90 per common share, except if we pay before the maturity all or any portion of the convertible debentures, in which case the applicable pro rata share of the contingent warrants will be exercisable for the remaining term of the convertible debentures at a fixed price of $1.90 per common share), (vi) warrants issued in connection with the our February 2017 public offering to purchase up to 1,904,034 common shares at an exercise price of $2.15 per common share, (vii) broker warrants issued in connection with our December 2017 public offering to purchase up to 495,050 common shares at an exercise price of US$1.26 per common share, and (viii) warrants issued in connection with our December 2017 public offering to purchase up to 9,802,935 common shares at an exercise price of US$1.26 per common share.
The following is a brief description of the rights, privileges, conditions and restrictions attaching to the common shares and preferred shares.
| - 74 - |
Common Shares
Voting Rights
Each common share entitles its holder to receive notice of, and to attend and vote at, all annual or special meetings of our shareholders. Each common share entitles its holder to one vote at any meeting of our shareholders, other than meetings at which only the holders of a particular class or series of shares are entitled to vote due to statutory provisions or the specific attributes of this class or series.
Dividends
Subject to the prior rights of the holders of preferred shares ranking before the common shares as to dividends, the holders of common shares are entitled to receive dividends as declared by the board our funds that are available for the payment of dividends.
Winding-up and Dissolution
In the event of our voluntary or involuntary winding-up or dissolution, or any other distribution of our assets among our shareholders for the purposes of winding up its affairs, the holders of common shares shall be entitled to receive, after payment by us to the holders of preferred shares ranking prior to common shares regarding the distribution of our assets in the case of winding-up or dissolution, share for share, the remainder of our property, with neither preference nor distinction. The order of priority, applicable to all classes of our shares with respect to the redemption, liquidation, dissolution or distribution of property (the order of priority) is as follows: First, the Class E non-voting shares; Second, the Class D non-voting shares; Third, the Class B multiple voting shares and Class C non-voting shares, pari passu; and Fourth, the common shares. Notwithstanding the order of priority, shareholders of a class of shares may renounce the order of priority by unanimous approval by all shareholders of that class of shares.
Preferred Shares
Class B Multiple Voting Shares
Each Class B multiple voting share entitles the holder thereof to 10 votes per share in all of our shareholder meetings.
Dividends. Holders of Class B multiple voting shares are entitled to receive, as and when such dividends are declared, an annual non-cumulative dividend of 5% on the amount paid for the said shares, payable at the time and in the manner which the directors may determine and subject to the order of priority.
Participation. Subject to the provisions of subsection 5.2.2 of our Articles, holders of Class B multiple voting shares do not have the right to participate in our profits or surplus assets.
Conversion. Holders of Class B multiple voting shares have the right, at their entire discretion, to convert, part or all of the Class B multiple voting shares they hold into common shares on the basis of 1 common share for each Class B multiple voting share converted.
Redemption. Subject to the provisions of the BCA and the order of priority, holders of Class B multiple voting shares have the right to demand from us, upon 30 days’ written notice, that we redeem the Class B multiple voting shares at a price equivalent to the amount paid for such shares plus the redemption premium, as defined in subsection 5.2.4.1 of the Articles, and any and all declared but yet unpaid dividends on same.
Liquidation. In the event of our dissolution or liquidation or any other distribution of our property, the Class B voting shareholders have the right to be reimbursed for the amount paid for their Class B multiple voting shares plus the redemption premium, as defined in subsection 5.2.4.1 of our Articles as well as the amount of any and all declared but yet unpaid dividends on their shares, subject to the order of priority.
Class C Non-Voting Shares
Subject to the provisions of the BCA, holders of Class C non-voting shares are neither entitled to vote at any meeting of our shareholders, receive a notice of any such meeting, nor attend any such meeting.
Dividends. Holders of Class C non-voting shares are entitled to receive, as and when such dividends are declared, an annual non-cumulative dividend of 5% on the amount paid for the said shares, plus a redemption premium as defined in subsection 5.3.6.1 of our Articles, payable at the time and in the manner which the directors may determine and subject to the order of priority.
Participation. Subject to the provisions of subsection 5.3.2 of our Articles, holders of Class C non-voting shares do not have the right to participate in our profits or surplus assets.
| - 75 - |
Conversion. Holders of Class C non-voting shares have the right, at their entire discretion, to convert, part or all of the Class C non-voting shares they hold into common shares on the basis of 1 common share for each Class C non-voting share converted.
Forced Conversion. All of our Class C non-voting shares shall automatically be converted in common shares upon the request of an unrelated third-party investor in us investing more than $500,000, or any other amount to be determined by the board of directors in us and requesting as a condition to the investment that the Class C non-voting shares be converted into common shares on the basis of 1 common share for each Class C non-voting share converted.
Redemption. Subject to the provisions of the BCA and the order of priority, holders of Class C non-voting shares have the right to demand, upon 30 days’ written notice, that we redeem their Class C non-voting shares at a price equivalent to the amount paid for the shares plus the redemption premium, as defined in subsection 5.3.6.1 of our Articles, and any and all declared but yet unpaid dividends on the shares.
Liquidation. In the event of our dissolution or liquidation or any other distribution of our property, Class C non-voting shareholders have the right to be reimbursed for the amount paid for their Class C non-voting shares plus the redemption premium, as defined in subsection 5.3.6.1 of our Articles, as well as the amount of any and all declared but yet unpaid dividends on their shares, subject to the order of priority.
Class D Non-Voting Shares
Subject to the provisions of the BCA, holders of Class D non-voting shares are neither entitled to vote at any meeting of the shareholders, receive a notice of any such meeting, nor attend any such meeting.
Dividends. Holders of Class D non-voting shares are entitled to receive, as and when such dividends are declared, a monthly non-cumulative dividend of 0.5% to 2% on the amount paid for the shares, plus a redemption premium as defined in subsection 5.4.6.1 of our Articles, payable at the time and in the manner which the directors may determine and subject to the order of priority.
Participation. Subject to the provisions of subsection 5.4.2 of our Articles, holders of Class D non-voting shares do not have the right to participate in our profits or surplus assets.
Conversion. Holders of Class D non-voting shares have the right, at their discretion, to convert, part or all of their Class D non-voting shares into common shares on the basis of a number of common shares equal to the number of Class D non-voting shares converted multiplied by a conversion ratio, calculated as follows:
The product obtained by multiplying a factor to be agreed at the time of the issuance of the Class D non-voting shares by the average amount paid per share for the Class D non-voting shares plus the redemption premium per share, as defined in subsection 5.4.6.1 of our Articles as well as the amount of any and all declared but yet paid
| Conversion Ratio | = | dividends on the shares |
| Fair market value of the common shares at the date of any conversion of Class D non-voting shares into common shares |
Conversion All of our Class D non-voting shares automatically convert into common shares upon the request of an unrelated third party investor in us, investing more than $500,000, or any other amount to be determined by the board of directors, in us and requesting as a condition to the investment that the Class D non-voting shares be converted into common shares in all cases, on the basis of a number of common shares equal to the number of Class D non-voting shares converted multiplied by the conversion ratio, calculated as follows:
The product obtained by multiplying a factor to be agreed at the time of the issuance of the Class D non-voting shares by the average amount paid per share for the Class D non-voting shares plus the redemption premium per share, as defined in subsection 5.4.6.1 of our Articles as well as the amount of any and all declared but yet paid
| Conversion Ratio | = | dividends on the shares |
| Fair market value of the common shares at the date of any conversion of Class D non-voting shares into common shares |
| - 76 - |
Redemption. Subject to the provisions of the BCA and the order of priority, holders of Class D non-voting shares have the right to demand, upon 30 days’ written notice, that we redeem their Class D non-voting shares at a price equivalent to the amount paid for the shares plus the redemption premium, as defined in subsection 5.4.6.1 of our Articles, and any and all declared but yet unpaid dividends on the shares.
Liquidation. In the event of our dissolution or liquidation or any other distribution of our property, the Class D non-voting shareholders shall have the right to be reimbursed for the amount paid for their Class D non-voting shares plus the redemption premium, as defined in subsection 5.4.6.1 of our Articles as well as the amount of any and all declared but yet unpaid dividends on their shares, subject to the order of priority.
Class E Non-Voting Shares
Subject to the provisions of the BCA, holders of Class E non-voting shares are neither entitled to vote at any meeting of the shareholders, receive a notice of any such meeting, nor attend any such meeting.
Dividends. Holders of Class E non-voting shares are entitled to receive, as and when such dividends are declared, a monthly non-cumulative dividend of 0.5% to 2% on the amount paid for the shares, payable at the time and in the manner which the directors may determine and subject to the order of priority.
Participation. Subject to the provisions of subsection 5.5.2 of our Articles, holders of Class E non-voting shares do not have the right to participate in our profits.
Conversion. Holders of Class E non-voting shares have the right, at their discretion, to convert, part or all of their Class E non-voting shares into common shares on the basis of a number of common shares equal to the number of Class E non-voting shares converted multiplied by the conversion ratio, calculated as follows:
| Conversion Ratio | = | The product obtained by multiplying a factor to be agreed at the time of the issuance of the Class E non-voting shares by the average amount paid per share for the Class E non-voting shares plus the amount of any and all declared but yet paid dividends on the shares |
| Fair market value of the common shares at the date of any conversion of Class E non-voting shares into common shares |
| - 77 - |
Redemption. Subject to the provisions of the BCA and the order of priority, we have the right, upon 30 days’ written notice, to redeem the Class E non-voting shares at a price equivalent to the amount paid for the shares and any and all declared but yet unpaid dividends on the shares.
Liquidation. In the event of our dissolution or liquidation or any other distribution of our property, the Class E non-voting shareholders have the right to be reimbursed for the amount paid for their Class E non-voting shares as well as the amount of any and all declared but yet unpaid dividends on the shares, subject to the order of priority.
Procedures to Change the Rights of Shareholders
In order to change the rights attached to all classes of our shares, the vote of at least 66 2/3% of the holders of each class, must be cast at a shareholders meeting called for amending the rights attached to our common shares or preferred shares, as the case may be.
Ordinary and Extraordinary Shareholders’ Meetings
Our by-laws provide that our annual meeting of shareholders must be held on a yearly basis on such date and on such time as may be fixed by the board. Our by-laws provide that special meetings of shareholders may be called at any time as determined by the board. Our shareholders are entitled to call special meetings of shareholders, provided that they hold at least 10% of the issued and outstanding shares entitled to vote at the meeting so called. Our by-laws provide that notice of each annual and special meeting of shareholders must be sent to the shareholders entitled to attend such meetings not less than 21 days and not more than 60 days before the date fixed for such meeting. Our by-laws provide that during any meeting of shareholders, the attendance, in person or by proxy, of at least two shareholders representing at least 10% of the issued and outstanding shares entitled to vote at the meeting will constitute a quorum.
Limitations on Rights to Own Securities
There exists no limitation on the right to own our securities.
Impediments to Change of Control
Neither our Articles nor by-laws contain any provision that would have an effect of delaying, deferring or preventing a change in control of us.
Stockholder Ownership Disclosure Threshold in Bylaws
Our Articles and By-laws do not contain any provision requiring a shareholder to disclose his ownership above a particular threshold.
| C. | Material Contracts |
For the two years preceding this annual report, we have not entered into any material contracts, other than contracts entered into in the ordinary course of our business, other than the indenture relating to the warrants that we issued in connection with our public offering of units in February 2017, the warrant agency agreement relating to the warrants that we issued in connection with our public offering of units in December 2017 and the underwriting agreement entered into in connection therewith and the indenture relating to the warrants that we issued in connection with our public offering of units in May 2018 and the underwriting agreement entered into in connection therewith.
| D. | Exchange Controls |
Subject to the following paragraph, there is no law or governmental decree or regulation in Canada that restricts the export or import of capital, or affects the remittance of dividends, interest or other payments to non-resident holders of our subordinate voting shares, other than withholding tax requirements.
There is no limitation imposed by Canadian law or by our Articles or our other charter documents on the right of a non-resident to hold or vote voting shares, other than as provided by the Investment Canada Act (Canada), or Investment Canada Act, the North American Free Trade Agreement Implementation Act (Canada), or North American Free Trade Agreement, and the World Trade Organization Agreement Implementation Act. The Investment Canada Act requires notification and, in certain cases, advance review and approval by the Government of Canada of an investment to establish a new Canadian business by a non-Canadian or of the acquisition by a “non-Canadian” of “control” of a “Canadian business”, all as defined in the Investment Canada Act. Generally, the threshold for review will be higher in monetary terms for a member of the World Trade Organization or North American Free Trade Agreement.
| - 78 - |
| E. | Taxation |
The following is a summary of certain U.S. federal income tax considerations to a U.S. Holder (as defined below) arising from and relating to the acquisition, ownership, and disposition of our common shares as capital assets.
This summary provides only general information and does not purport to be a complete analysis or listing of all potential U.S. federal income tax consequences that may apply to a U.S. Holder as a result of the acquisition, ownership, and disposition of our common shares. In addition, this summary does not take into account the individual facts and circumstances of any particular U.S. Holder that may affect the U.S. federal income tax consequences applicable to that U.S. Holder. Accordingly, this summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. Each U.S. Holder should consult its own tax advisor regarding the U.S. federal, U.S. state and local, and non-U.S. tax consequences arising from or relating to the acquisition, ownership, and disposition of our common shares.
No legal opinion from U.S. legal counsel or ruling from the Internal Revenue Service, or IRS, has been requested, or will be obtained, regarding the U.S. federal income tax consequences to U.S. Holders of the acquisition, ownership, and disposition of our common shares. This summary is not binding on the IRS, and the IRS is not precluded from taking a position that is different from, and contrary to, the positions taken in this summary. In addition, because the authorities on which this summary is based are subject to various interpretations, the IRS and the U.S. courts could disagree with one or more of the positions taken in this summary.
Scope of this Disclosure
Authorities
This summary is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, U.S. Treasury Regulations promulgated thereunder (whether final, temporary or proposed), published IRS rulings, judicial decisions, published administrative positions of the IRS, and the Convention between Canada and the United States of America with Respect to Taxes on Income and on Capital, signed September 26, 1980, as amended (the Canada-U.S. Tax Treaty). Any of the authorities on which this summary is based could be changed in a material and adverse manner at any time, and any such change could be applied on a retroactive basis. Unless otherwise discussed, this summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation.
U.S. Holders
For purposes of this summary, a “U.S. Holder” is a beneficial owner of common shares that, for U.S. federal income tax purposes, is (a) an individual who is a citizen or resident of the United States, (b) a corporation, or other entity classified as a corporation for U.S. federal income tax purposes, that is created or organized in or under the laws of the U.S., any state in the United States or the District of Columbia, (c) an estate if the income of such estate is subject to U.S. federal income tax regardless of the source of such income, or (d) a trust if (i) such trust has validly elected to be treated as a U.S. person for U.S. federal income tax purposes or (ii) a U.S. court is able to exercise primary supervision over the administration of such trust and one or more U.S. persons have the authority to control all substantial decisions of such trust.
U.S. Holders Subject to Special U.S. Federal Income Tax Rules Not Addressed
This summary does not address the U.S. federal income tax consequences applicable to U.S. Holders that are subject to special provisions under the Code, including, but not limited to, the following U.S. Holders: (a) U.S. Holders that are tax-exempt organizations, qualified retirement plans, individual retirement accounts, or other tax deferred accounts; U.S. Holders that are financial institutions, insurance companies, real estate investment trusts, or regulated investment companies; U.S. Holders that are dealers in securities or currencies or U.S. Holders that are traders in securities that elect to apply a mark-to-market accounting method; (d) U.S. Holders that have a “functional currency” other than the U.S. dollar; (e) U.S. Holders subject to the alternative minimum tax provisions of the Code; (f) U.S. Holders that own common shares as part of a straddle, hedging transaction, conversion transaction, integrated transaction, constructive sale, or other arrangement involving more than one position; (g) U.S. Holders that acquired common shares through the exercise of employee stock options or otherwise as compensation for services; (h) U.S. Holders that hold common shares other than as a capital asset within the meaning of Section 1221 of the Code; (i) U.S. Holders that beneficially own (directly, indirectly or by attribution) 10% or more of our voting securities or otherwise held 10% or more of our total combined voting power; and (j) U.S. expatriates. U.S. Holders that are subject to special provisions under the Code, including U.S. Holders described above, should consult their own tax advisor regarding the U.S. federal, U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences arising from and relating to the acquisition, ownership, and disposition of the common shares.
If an entity or arrangement that is classified as a partnership for U.S. federal income tax purposes holds common shares, the U.S. federal income tax consequences to that partnership and the partners of that partnership generally will depend on the activities of the partnership and the status of the partners. Partners of entities that are classified as partnerships for U.S. federal income tax purposes should consult their own tax advisors regarding the U.S. federal income tax consequences arising from and relating to the acquisition, ownership and disposition of the common shares.
| - 79 - |
Tax Consequences Other than U.S. Federal Income Tax Consequences Not Addressed
This summary does not address the U.S. estate and gift, alternative minimum, state, local or non-U.S. tax consequences to U.S. Holders of the acquisition, ownership, and disposition of our common shares. Each U.S. Holder should consult its own tax advisor regarding the U.S. estate and gift, alternative minimum, state, local and foreign tax consequences arising from and relating to the acquisition, ownership, and disposition of our common shares.
U.S. Federal Income Tax Considerations of the Acquisition, Ownership, and Disposition of Common Shares
Distributions on Common Shares
Subject to the possible application of the passive foreign investment company, or PFIC, rules described below (see the more detailed discussion below at “Passive Foreign Investment Company Rules”), a U.S. Holder that receives a distribution, including a constructive distribution or a taxable stock distribution, with respect to the common shares generally will be required to include the amount of that distribution in gross income as a dividend (without reduction for any Canadian income tax withheld from such distribution) to the extent of our current or accumulated “earnings and profits” (as computed for U.S. federal income tax purposes). To the extent that a distribution exceeds our current and accumulated “earnings and profits”, the excess amount will be treated (a) first, as a tax-free return of capital to the extent of a U.S. Holder’s adjusted tax basis in the common shares with respect to which the distribution is made (resulting in a corresponding reduction in the tax basis of those common shares) and, (b) thereafter, as gain from the sale or exchange of those common shares (see the more detailed discussion at “—Disposition of Common Shares” below). We do not intend to calculate our current or accumulated earnings and profits for U.S. federal income tax purposes and, therefore, will not be able to provide Holders with that information. U.S. Holders should therefore assume that any distribution by us with respect to our common shares will constitute a dividend. However, U.S. Holders should consult their own tax advisors regarding whether distributions from us should be treated as dividends for U.S. federal income tax purposes. Dividends paid on our common shares generally will not be eligible for the “dividends received deduction” allowed to corporations under the Code with respect to dividends received from U.S. corporations.
A dividend paid by us generally will be taxed at the preferential tax rates applicable to long-term capital gains if, among other requirements, (a) we are a “qualified foreign corporation” (as defined below), (b) the U.S. Holder receiving the dividend is an individual, estate, or trust, and (c) the dividend is paid on common shares that have been held by the U.S. Holder for at least 61 days during the 121-day period beginning 60 days before the “ex-dividend date” (i.e., the first date that a purchaser of the common shares will not be entitled to receive the dividend).
For purposes of the rules described in the preceding paragraph, we generally will be a “qualified foreign corporation”, or a QFC, if (a) we are eligible for the benefits of the Canada-U.S. Tax Treaty, or (b) our common shares are readily tradable on an established securities market in the United States, within the meaning provided in the Code. However, even we satisfy one or more of the requirements, we will not be treated as a QFC if we are classified as a PFIC (as discussed below) for the taxable year during which we pay the applicable dividend or for the preceding taxable year. The dividend rules are complex, and each U.S. Holder should consult its own tax advisor regarding the application of those rules to them in their particular circumstances. Even if we satisfy one or more of the requirements, as noted below, there can be no assurance that we will not become a PFIC in the future. Thus, there can be no assurance that we will qualify as a QFC.
Disposition of Common Shares
Subject to the possible application of the PFIC rules described below (see more detailed discussion below at “Passive Foreign Investment Company Rules”), a U.S. Holder will recognize gain or loss on the sale or other taxable disposition of common shares (that is treated as a sale or exchange for U.S. federal income tax purposes) equal to the difference, if any, between (a) the U.S. dollar value of the amount realized on the date of the sale or disposition and (b) the U.S. Holder’s adjusted tax basis (determined in U.S. dollars) in the common shares sold or otherwise disposed of. Any such gain or loss generally will be capital gain or loss, which will be long-term capital gain or loss if the common shares are held for more than one year. Each U.S. Holder should consult its own tax advisor as to the tax treatment of dispositions of common shares in exchange for Canadian dollars.
Preferential tax rates apply to long-term capital gains of a U.S. Holder that is an individual, estate, or trust. There are currently no preferential tax rates for long-term capital gains of a U.S. Holder that is a corporation. Deductions for capital losses are subject to complex limitations.
| - 80 - |
Passive Foreign Investment Company Rules
Special, generally unfavorable, rules apply to the ownership and disposition of the stock of a PFIC. For U.S. federal income tax purposes, a non-U.S. corporation is classified as a PFIC for each taxable year in which either:
| · | at least 75% of its gross income is “passive” income (referred to as the “income test”); or |
| · | at least 50% of the average value of its assets is attributable to assets that produce passive income or are held for the production of passive income (referred to as the “asset test”). |
Passive income includes the following types of income:
| · | dividends, royalties, rents, annuities, interest, and income equivalent to interest; and |
| · | net gains from the sale or exchange of property that gives rise to dividends, interest, royalties, rents, or annuities and certain gains from the commodities transactions. |
In determining whether we are a PFIC, we will be required to take into account a pro rata portion of the income and assets of each corporation in which we own, directly or indirectly, at least 25% by value.
We have not made a determination as to whether we were a PFIC for the 2017 taxable year(s) or whether we will be a PFIC for the current taxable year. Accordingly, there can be no assurance that we were not a PFIC for the 2017 taxable year(s). Whether we are a PFIC depends on complex U.S. federal income tax rules that are subject to differing interpretations and whose application to us is uncertain. Further, since our PFIC status will depend upon the composition of our income and assets and the fair market value of our assets from time to time (including whether we own, directly or indirectly, at least 25% by value, of the stock of any subsidiary) and generally cannot be determined until the end of a taxable year, there can be no assurance that we will not be a PFIC for the current taxable year. In addition, we cannot predict whether the composition of our income and assets (including income and assets held indirectly) or the fair market value of its assets from time to time may result in it being treated as a PFIC in any future taxable year. Accordingly, no assurance can be given that we are not a PFIC or will not become a PFIC in subsequent taxable years.
Generally, if we are or have been treated as a PFIC for any taxable year during a U.S. Holder’s holding period of common shares, any “excess distribution” with respect to the common shares would be allocated rateably over the U.S. Holder’s holding period. The amounts allocated to the taxable year of the excess distribution and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations in that taxable year, as appropriate, and an interest charge would be imposed on the amount allocated to that taxable year. Distributions made in respect of common shares during a taxable year will be excess distributions to the extent they exceed 125% of the average of the annual distributions on common shares received by the U.S. Holder during the preceding three taxable years or the U.S. Holder’s holding period, whichever is shorter.
Generally, if we are treated as a PFIC for any taxable year during which a U.S. Holder owns common shares, any gain on the disposition of the common shares would be treated as an excess distribution and would be allocated rateably over the U.S. Holder’s holding period and subject to taxation in the same manner as described in the preceding paragraph.
Certain elections may be available (including a “mark-to-market” or “qualified electing fund” election) to U.S. Holders in limited circumstances that may mitigate the adverse consequences resulting from PFIC status, particularly if they are made in the first taxable year during such holder’s holding period in which we are treated as a PFIC. U.S. Holders should be aware that, for each tax year, if any, that we are a PFIC, we can provide no assurances that we will make available to U.S. Holders the information U.S. Holders require to make a “qualified electing fund” election with respect to us.
If we were to be treated as a PFIC in any taxable year, a U.S. Holder will generally be required to file an annual report with the IRS containing such information as the U.S. Treasury Department may require.
Each current or prospective U.S. Holder should consult its own tax advisor regarding our status as a PFIC, the possible effect of the PFIC rules to such holder and information reporting required if we were a PFIC, as well as the availability of any election that may be available to the holder to mitigate adverse U.S. federal income tax consequences of holding shares in a PFIC.
Receipt of Foreign Currency
The amount of a distribution paid in Canadian dollars or Canadian dollar proceeds received on the sale or other taxable disposition of common shares will generally be equal to the U.S. dollar value of the currency on the date of receipt. If any Canadian dollars received with respect to the common shares are later converted into U.S. dollars, U.S. Holders may realize gain or loss on the conversion. Any gain or loss generally will be treated as ordinary income or loss and generally will be from sources within the United States for U.S. foreign tax credit purposes. Each U.S. Holder should consult its own tax advisor concerning the possibility of foreign currency gain or loss if any such currency is not converted into U.S. dollars on the date of receipt.
| - 81 - |
Foreign Tax Credit
Subject to certain limitations, a U.S. Holder who pays (whether directly or through withholding) Canadian or other foreign income tax with respect to the common shares may be entitled, at the election of the U.S. Holder, to receive either a deduction or a credit for Canadian or other foreign income tax paid. Dividends paid on common shares generally will constitute income from sources outside the United States. The foreign tax credit rules (including the limitations with respect thereto) are complex, and each U.S. Holder should consult its own tax advisor regarding the foreign tax credit rules, having regard to such holder’s particular circumstances.
Information Reporting; Backup Withholding
Generally, information reporting and backup withholding will apply to distributions on, and the payment of proceeds from the sale or other taxable disposition of, the common shares unless (i) the U.S. Holder is a corporation or other exempt entity, or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that the U.S. Holder is not subject to backup withholding.
Backup withholding is not an additional tax. Any amount withheld generally will be creditable against a U.S. Holder’s U.S. federal income tax liability or refundable to the extent that it exceeds such liability provided the required information is provided to the IRS in a timely manner.
In addition, certain categories of U.S. Holders must file information returns with respect to their investment in a non-U.S. corporation. For example, certain U.S. Holders must file IRS Form 8938 with respect to certain “specified foreign financial assets” (such as the common shares) with an aggregate value in excess of US$50,000 (and, in some circumstances, a higher threshold). Failure to do so could result in substantial penalties and in the extension of the statute of limitations with respect to such holder’s U.S. federal income tax returns. Each U.S. Holder should consult its own tax advisor regarding application of the information reporting and backup withholding rules to it in connection with an investment in our common shares.
Medicare Contribution Tax
U.S. Holders that are individuals, estates or certain trusts generally will be subject to a 3.8% Medicare contribution tax on, among other things, dividends on, and capital gains from the sale or other taxable disposition of, common shares, subject to certain limitations and exceptions. Each U.S. Holder should consult its own tax advisor regarding possible application of this additional tax to income earned in connection with an investment in our common shares.
| F. | Dividends and Paying Agents |
Not applicable.
| G. | Statement by Experts |
Not applicable.
| H. | Documents on Display |
Any statement in this annual report about any of our contracts or other documents is not necessarily complete. If the contract or document is filed as an exhibit to this annual report, the contract or document is deemed to modify the description contained in this annual report. You must review the exhibits themselves for a complete description of the contract or document.
Our SEC filings are available at the SEC’s website at www.sec.gov. You may also read and copy any document we file with the SEC at the public reference facilities maintained by the SEC at SEC Headquarters, Public Reference Section, 100 F Street, N.E., Washington D.C. 20549. You may obtain information on the operation of the SEC’s public reference facilities by calling the SEC at 1-800-SEC-0330. In addition, we are required by Canadian securities laws to file documents electronically with Canadian securities regulatory authorities and these filings are available on our SEDAR profile at www.sedar.com. Requests for such documents should be directed to our Corporate Secretary.
| I. | Subsidiary Information |
Not applicable.
| - 82 - |
| Item 11. | Quantitative and Qualitative Disclosure about Market Risk |
Information relating to quantitative and qualitative disclosures about market risks is detailed in “Item 5. Operating and Financial Review and Prospects”, as well as in Note 19 to our audited financial statements contained in “Item 17. Financial Statements”.
| Item 12. | Description of Securities other than Equity Securities |
| A. | Debt Securities |
Not applicable.
| B. | Warrants and Rights |
Not applicable.
| C. | Other Securities |
Not applicable.
| D. | American Depositary Shares |
Not applicable.
| - 83 - |
| Item 13. | Defaults, Dividend Arrearages and Delinquencies |
None.
| Item 14. | Material Modification to the Rights of Security Holdings and Use of Proceeds |
None.
| Item 15. | Controls and Procedures |
Disclosure Controls and Procedures
As of the end of the period covered by this annual report, our management, with the participation of our CEO and CFO, has performed an evaluation of the effectiveness of our disclosure controls and procedures within the meaning of Rules 13a-15 (e) and 15d-15(e) of the Exchange Act. Based upon this evaluation, our management has concluded that, as of March 31, 2018, our existing disclosure controls and procedures were effective. It should be noted that while the CEO and CFO believe that our disclosure controls and procedures provide a reasonable level of assurance that they are effective, they do not expect the disclosure controls and procedures to be capable of preventing all errors and fraud. A control system, no matter how well conceived or operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
Management’s Report on Internal Controls over Financial Reporting
Our management, with the participation of our CEO and CFO, is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control system was designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation and fair presentation of its published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective may not prevent or detect misstatements and can provide only reasonable assurance with respect to financial statement preparation and presentation. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Our management conducted an assessment of the design and operation effectiveness of our internal control over financial reporting as of March 31, 2018. In making this assessment, we used the criteria established within the Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this assessment, our management has concluded that, as of March 31, 2018, our internal control over financial reporting was effective.
Changes in Internal Control over Financial Reporting
No changes were made to our internal controls over financial reporting that occurred during the quarter and fiscal year ended March 31, 2018 that have materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
We qualify as an “emerging growth company” under Section 3(a)(80) of the Exchange Act, as a result of enactment of the Jumpstart Our Business Startups Act of 2012, or JOBS Act. Under the JOBS Act, emerging growth companies are exempt from Section 404(b) of the Sarbanes-Oxley Act of 2002, which generally requires that a public company’s registered public accounting firm provide an attestation report relating to management’s assessment of internal control over financial reporting. We qualify as an emerging growth company and therefore have not included in, or incorporated by reference into, this annual report such an attestation report as of the end of the period covered by this annual report.
| Item 16. | Reserved |
| Item 16A. | Audit Committee Financial Expert |
Our board of directors has determined that Mr. Canan is the “audit committee financial expert”, as defined by applicable regulations of the Commission. The Commission has indicated that the designation of Mr. Canan as an audit committee financial expert does not make him an “expert” for any purpose, impose any duties, obligations or liability on Mr. Canan that are greater than those imposed on members of the audit committee and board of directors who do not carry this designation or affect the duties, obligations or liability of any other member of the audit committee or board of directors.
| - 84 - |
| Item 16B. | Code of Ethics |
The board of directors adopted a Code of Business Conduct and Ethics for our directors, officers and employees on May 31, 2007, which can be found on SEDAR at www.sedar.com and on our web site on www.acastipharma.com. A copy of the Code of Ethics and Conduct can also be obtained by contacting our Corporate Secretary. Any breach of the Code of Ethics must be brought to the attention of the board of directors by our CEO or other senior executive officer. No report has ever been filed which pertains to any conduct of a director or executive officer that constitutes a breach of the Code of Business Conduct and Ethics.
The board of directors also adopted an insider trading program for its directors, officers and employees and adopted recently a majority voting policy for the election of proposed director candidates at our annual general shareholders meeting.
| Item 16C. | Principal Accountant Fees and Services Audit Fees |
“Audit fees” consist of fees for professional services for the audit of our annual financial statements, interim reviews and limited procedures on interim financial statements, securities filings and consultations on accounting or disclosure issues. KPMG LLP, our external auditors, billed $349,100 for audit fees for the fiscal year ended March 31, 2018 and $235,400 for audit fees for the fiscal year ended March 31, 2017.
Audit-Related Fees
“Audit-related fees” consist of fees for professional services that are reasonably related to the performance of the audit or review of our financial statements and which are not reported under “Audit Fees” above. KPMG LLP billed $8,440 for the fiscal year ended March 31, 2018 and $6,550 for the fiscal year ended March 31, 2017.
Tax Fees
“Tax fees” consist of fees for professional services for tax compliance, tax advice and tax planning. KPMG LLP billed $57,100 and $31,600 for tax fees for fiscal year ended March 31, 2017. Tax fees include, but are not limited to, preparation of tax returns.
All Other Fees
“Other fees” include all other fees billed for professional services other than those mentioned hereinabove. KPMG LLP billed no fees under this category for the fiscal years ended March 31, 2018 and March 31, 2017.
Pre-Approval Policies and Procedures
The audit committee approves all audit, audit-related services, tax services and other non-audit related services provided by the external auditors in advance of any engagement. Under the Sarbanes-Oxley Act of 2002, audit committees are permitted to approve certain fees for non-audit related services pursuant to a de minimus exception prior to the completion of an audit engagement. Non-audit related services satisfy the de minimus exception if the following conditions are met:
| · | the aggregate amount of all non-audit services that were not pre-approved is reasonably expected to constitute no more than five per cent of the total amount of fees paid by us and our subsidiaries to our external auditors during the fiscal year in which the services are provided; |
| · | we or our subsidiaries, as the case may be, did not recognize the services as non-audit services at the time of the engagement; and |
| · | the services are promptly brought to the attention of the audit committee and approved, prior to the completion of the audit, by the audit committee or by one or more of its members to whom authority to grant such approvals had been delegated by the audit committee. |
None of the services described above under “Principal Accountant Fees and Services” were approved by the audit committee pursuant to the de minimus exception.
| Item 16D. | Exemptions from the Listing Standards for Audit Committees |
Not applicable.
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers |
Not applicable.
| Item 16F. | Change in Registrant’s Certifying Accountant |
None.
| - 85 - |
| Item 16G. | Corporation Governance |
NASDAQ Marketplace Rule 5615(a)(3) permits a foreign private issuer to follow its home country practice in lieu of certain of the requirements of the Rule 5600 Series. A foreign private issuer that follows a home country practice in lieu of one or more provisions of the Rule 5600 Series is required to disclose in its annual report filed with the SEC, or on its website, each requirement of the Rule 5600 Series that it does not follow and describe the home country practice followed by the issuer in lieu of such NASDAQ corporate governance requirements. We do not follow NASDAQ Marketplace Rule 5620(c), but instead follow our home country practice. The NASDAQ minimum quorum requirement under Rule 5620(c) for a meeting of shareholders is 33.33% of the outstanding shares of common voting stock. Our quorum requirement, as set forth in our by-laws, is that a quorum for a meeting of our holders of common shares is the attendance, in person or by proxy, of the shareholders representing 10% of our common shares. The foregoing is consistent with the laws, customs and practices in Québec, Canada, and the rules and policies of the TSX-V.
| Item 16H. | Mining Safety Disclosure |
Not applicable.
| Item 17. | Financial Statements |
The financial statements of Acasti Pharma Inc. are located at the end of this annual report, beginning on page F-1.
| Item 18. | Financial Statements |
See Item 17.
| Item 19. | Exhibits |
| - 86 - |
| 101.INS* | XBRL Instance Document |
| 101.SCH* | XBRL Taxonomy Extension Schema Document |
| 101.CAL* | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF* | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB* | XBRL Taxonomy Extension Labels Linkbase Document |
| 101.PRE* | XBRL Taxonomy Extension Presentation Linkbase Document |
* Filed herewith.
| - 87 - |
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on this Annual Report and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
| ACASTI PHARMA INC. | |||
| By: | /s/ Janelle D’Alvise | ||
| Name: | Janelle D’Alvise | ||
| Title: | Principal Executive Officer | ||
| Date: June 29, 2018 | |||
| - 88 - |
Financial Statements of
Acasti pharma inc.
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| F-1 |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Acasti Pharma Inc.
Opinion on the Financial Statements
We have audited the accompanying statements of financial position of Acasti Pharma Inc. (the "Company") as of March 31, 2018, and 2017, the related statements of earnings and comprehensive loss, changes in equity and cash flows for the periods ended March 31, 2018, March 31, 2017 and February 29, 2016, and the related notes (collectively referred to as the "financial statements").
In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2018, and 2017, and its financial performance and its cash flows for the periods ended March 31, 2018, March 31, 2017 and February 29, 2016, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Material Uncertainty Related to Going Concern
Without qualifying our opinion on the financial statements, we draw attention to Note 2 (c) to the financial statements, which indicates that the Company has incurred operating losses and negative cash flows from operations since inception, the Company’s current assets are projected to be significantly less than what will be needed, and the Company needs to obtain additional financing. As stated in Note 2 (c) to the financial statements, these events or conditions, along with other matters as set forth in Note 2 (c), indicate that a material uncertainty exists that casts substantial doubt on the Company’s ability to continue as a going concern.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits.
We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB, and in accordance with the ethical requirements that are relevant to our audit of the financial statements in Canada.

| F-2 |
![]()
Page 2
We conducted our audits in accordance with Canadian generally accepted auditing standards and the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purposes of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Other Matter
The financial statements of Acasti Pharma Inc. as at February 28, 2017 and for the twelve-month and one-month periods ended February 28, 2017 and March 31, 2017, respectively, are unaudited. Accordingly, we do not express an opinion on them.
We have served as Company’s auditor since 2009.
June 27, 2018
Montréal, Canada
*CPA auditor, CA, public accountancy permit No. A122596
| F-3 |
Acasti pharma inc.
Financial Statements
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Financial Statements | ||
| Statements of Financial Position | F-5 | |
| Statements of Earnings and Comprehensive Loss | F-6 | |
| Statements of Changes in Equity | F-7 | |
| Statements of Cash Flows | F-10 | |
| Notes to Financial Statements | F-11 | |
| F-4 |
Acasti Pharma inc.
Statements of Financial Position
As at March 31, 2018, March 31, 2017 and February 28, 2017
| March 31, 2018 | March 31, 2017 | February 28, 2017 | ||||||||||||
| (Unaudited) | ||||||||||||||
| (thousands of Canadian dollars) | Notes | $ | $ | $ | ||||||||||
| Assets | ||||||||||||||
| Current assets: | ||||||||||||||
| Cash and cash equivalents | 23 | 8,223 | 9,772 | 10,573 | ||||||||||
| Receivables | 4 | 759 | 206 | 166 | ||||||||||
| Other Assets | 5 | 104 | — | — | ||||||||||
| Prepaid expenses | 406 | 303 | 270 | |||||||||||
| Total current assets | 9,492 | 10,281 | 11,009 | |||||||||||
| Marketable securities | 23 | 26 | — | — | ||||||||||
| Other Asset | 5 | 555 | — | — | ||||||||||
| Equipment | 8 | 2,821 | 2,787 | 2,776 | ||||||||||
| Intangible assets | 9 | 10,065 | 12,388 | 12,582 | ||||||||||
| Total assets | 22,959 | 25,456 | 26,367 | |||||||||||
| Liabilities and Equity | ||||||||||||||
| Current liabilities | ||||||||||||||
| Trade and other payables | 10 | 6,697 | 2,138 | 2,405 | ||||||||||
| Total current liabilities | 6,697 | 2,138 | 2,405 | |||||||||||
| Derivative warrant liabilities | 11, 13(e) | 6,426 | 209 | 187 | ||||||||||
| Unsecured convertible debentures | 12 | 1,612 | 1,406 | 1,389 | ||||||||||
| Total liabilities | 14,735 | 3,753 | 3,981 | |||||||||||
| Equity: | ||||||||||||||
| Share capital | 13 | 73,338 | 66,576 | 66,576 | ||||||||||
| Other equity | 13 | 309 | 309 | 309 | ||||||||||
| Contributed surplus | 6,956 | 5,693 | 5,607 | |||||||||||
| Deficit | (72,379 | ) | (50,875 | ) | (50,106 | ) | ||||||||
| Total equity | 8,224 | 21,703 | 22,386 | |||||||||||
| Commitments and contingencies | 21 | |||||||||||||
| Total liabilities and equity | 22,959 | 25,456 | 26,367 |
See accompanying notes to financial statements.
On behalf of the Board:
| /s/ Dr. Roderick Carter | /s/Jean-Marie Canan |
| Roderick Carter | Jean-Marie Canan |
| Chair of the Board | Director |
| F-5 |
Acasti pharma INC.
Statements of Earnings and Comprehensive Loss
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Thirteen- months ended |
Month ended |
Twelve- months ended |
||||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 |
February 28, 2017 |
February 29, 2016 |
||||||||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||||||||
| (thousands of Canadian dollars, except per share data) | Notes | $ | $ | $ | $ | $ | ||||||||||||||||
| Research and development expenses, net of government assistance | 7 | (15,669 | ) | (7,653 | ) | (426 | ) | (7,227 | ) | (7,566 | ) | |||||||||||
| General and administrative expenses | (4,027 | ) | (3,557 | ) | (292 | ) | (3,265 | ) | (2,046 | ) | ||||||||||||
| Loss from operating activities | (19,696 | ) | (11,210 | ) | (718 | ) | (10,492 | ) | (9,612 | ) | ||||||||||||
| Change in fair value of warrant liabilities | 11,15 | (344 | ) | (53 | ) | (22 | ) | (31 | ) | 2,201 | ||||||||||||
| Other financial expenses | 13 (b),15 | (1,464 | ) | (113 | ) | (29 | ) | (84 | ) | 1,094 | ||||||||||||
| Net financial expenses | (1,808 | ) | (166 | ) | (51 | ) | (115 | ) | 3,295 | |||||||||||||
| Net loss and comprehensive loss before income tax | (21,504 | ) | (11,376 | ) | (769 | ) | (10,607 | ) | (6,317 | ) | ||||||||||||
| Deferred income tax recovery | — | 129 | — | 129 | — | |||||||||||||||||
| Net loss and total comprehensive loss | (21,504 | ) | (11,247 | ) | (769 | ) | (10,478 | ) | (6,317 | ) | ||||||||||||
| Basic and diluted loss per share | 17 | (1.23 | ) | (1.01 | ) | (0.05 | ) | (0.97 | ) | (0.59 | ) | |||||||||||
| Weighted average number of shares outstanding | 17,486,515 | 11,094,512 | 14,702,556 | 10,788,075 | 10,659,936 |
See accompanying notes to financial statements
| F-6 |
Acasti pharma INC.
Statements of Changes in Equity
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Share capital | Other | Contributed | ||||||||||||||||||||||||
| Number | Dollar | equity | surplus | Deficit | Total | |||||||||||||||||||||
| (thousands of Canadian dollars) | Notes | $ | $ | $ | $ | $ | ||||||||||||||||||||
| Balance, March 31, 2017 | 14,702,556 | 66,576 | 309 | 5,693 | (50,875 | ) | 21,703 | |||||||||||||||||||
| Net loss and total comprehensive loss for the period | — | — | — | — | (21,504 | ) | (21,504 | ) | ||||||||||||||||||
| 14,702,556 | 66,576 | 309 | 5,693 | (72,379 | ) | 199 | ||||||||||||||||||||
| Transactions with owners, recorded directly in equity | ||||||||||||||||||||||||||
| Contributions by and distributions to equity holders | ||||||||||||||||||||||||||
| Public offering | 13 | 10,667,169 | 6,169 | — | 406 | — | 6,575 | |||||||||||||||||||
| Warrants exercised | 178,721 | 456 | — | (72 | ) | — | 384 | |||||||||||||||||||
| Share-based payment transactions | 16 | — | — | — | 929 | — | 929 | |||||||||||||||||||
| Issuance of shares for payment of interest on convertible debentures | 13(d) | 89,769 | 137 | — | — | 137 | ||||||||||||||||||||
| Total contributions by and distributions to equity holders | 10,935,659 | 6,762 | — | 1,263 | — | 8,025 | ||||||||||||||||||||
| Balance at March 31, 2018 | 25,638,215 | 73,338 | 309 | 6,956 | (72,379 | ) | 8,224 | |||||||||||||||||||
See accompanying notes to financial statements.
| F-7 |
Acasti pharma INC.
Statements of Changes in Equity, Continued
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Share capital | Other | Contributed | ||||||||||||||||||||||||
| Number | Dollar | equity | surplus | Deficit | Total | |||||||||||||||||||||
| (thousands of Canadian dollars) | Notes | $ | $ | $ | $ | $ | ||||||||||||||||||||
| Balance, February 29, 2016 | 10,712,038 | 61,973 | — | 4,875 | (39,628 | ) | 27,220 | |||||||||||||||||||
| Net loss and total comprehensive loss for the twelve-month period (unaudited) | — | — | — | — | (10,478 | ) | (10,478 | ) | ||||||||||||||||||
| Net loss and total comprehensive loss for the one-month period (unaudited) | — | — | — | — | (769 | ) | (769 | ) | ||||||||||||||||||
| Net loss and total comprehensive loss for the thirteen-month period | — | — | — | — | (11,247 | ) | (11,247 | ) | ||||||||||||||||||
| 10,712,038 | 61,973 | — | 4,875 | (50,875 | ) | 15,973 | ||||||||||||||||||||
| Transactions with owners, recorded directly in equity | ||||||||||||||||||||||||||
| Contributions by and distributions to equity holders | ||||||||||||||||||||||||||
| Public offering | 13(c) | 3,930,518 | 4,509 | — | 144 | — | 4,653 | |||||||||||||||||||
| Issue of unsecured convertible debentures, net of deferred income tax expense of $129 income tax expense of $129 | 13,19 | — | — | 309 | — | — | 309 | |||||||||||||||||||
| Equity settled non-employee share-based payment | 60,000 | 94 | — | — | — | 94 | ||||||||||||||||||||
| Share-based payment transactions for the twelve-month period (unaudited) | 16 | — | — | — | 588 | — | 588 | |||||||||||||||||||
| Share-based payment transactions for the one-month period (unaudited) | 16 | — | — | — | 86 | — | 86 | |||||||||||||||||||
| Share-based payment transactions for the thirteen-month period | 16 | — | — | — | 674 | — | 674 | |||||||||||||||||||
| Total contributions by and distributions to equity holders for the twelve-month period (unaudited) | 3,990,518 | 4,603 | 309 | 732 | — | 5,644 | ||||||||||||||||||||
| Total contributions by and distributions to equity holders for the one-month period (unaudited) | — | — | — | 86 | — | 86 | ||||||||||||||||||||
| Total contributions by and distributions to equity holders for the thirteen-month period | 3,990,518 | 4,603 | 309 | 818 | — | 5,730 | ||||||||||||||||||||
| Balance at February 28, 2017 (unaudited) | 14,702,556 | 66,576 | 309 | 5,607 | (50,106 | ) | 22,386 | |||||||||||||||||||
| Balance at March 31, 2017 | 14,702,556 | 66,576 | 309 | 5,693 | (50,875 | ) | 21,703 | |||||||||||||||||||
See accompanying notes to financial statements.
| F-8 |
Acasti pharma INC.
Statements of Changes in Equity, Continued
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Share capital | Other | Contributed | ||||||||||||||||||||||||
| Notes | Amount | Dollar | equity | surplus | Deficit | Total | ||||||||||||||||||||
| (thousands of Canadian dollars) | $ | $ | $ | $ | $ | |||||||||||||||||||||
| Balance, February 28, 2015 | 10,644,440 | 61,628 | — | 4,911 | (33,311 | ) | 33,228 | |||||||||||||||||||
| Net loss and total comprehensive loss for the year | — | — | — | — | (6,317 | ) | (6,317 | ) | ||||||||||||||||||
| 10,644,440 | 61,628 | — | 4,911 | (39,628 | ) | 26,911 | ||||||||||||||||||||
| Transactions with owners, recorded directly in equity | ||||||||||||||||||||||||||
| Contributions by and distributions to equity holders | ||||||||||||||||||||||||||
| Share-based payment transactions | 16 | — | — | — | 309 | — | 309 | |||||||||||||||||||
| Issuance of shares | 13(c) | 50,000 | 101 | — | (102 | ) | — | (1 | ) | |||||||||||||||||
| Share options exercised | 16 | 250 | 1 | — | — | — | 1 | |||||||||||||||||||
| RSUs released | 17,348 | 243 | — | (243 | ) | — | — | |||||||||||||||||||
| Total contributions by and distributions to equity holders | 67,598 | 345 | — | (36 | ) | — | 309 | |||||||||||||||||||
| Balance at February 29, 2016 | 10,712,038 | 61,973 | — | 4,875 | (39,628 | ) | 27,220 | |||||||||||||||||||
| F-9 |
acasti pharma INC.
Statements of Cash Flows
For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 and year ended February 29, 2016
| Thirteen-months ended |
Month ended |
Twelve-months ended |
||||||||||||||||||||
| Notes | March 31, 2018 |
March 31, 2017 |
March 31, 2017 (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
|||||||||||||||||
| (thousands of Canadian dollars) | $ | $ | $ | $ | $ | |||||||||||||||||
| Cash flows used in operating activities: | ||||||||||||||||||||||
| Net loss for the period | (21,504 | ) | (11,247 | ) | (769 | ) | (10,478 | ) | (6,317 | ) | ||||||||||||
| Adjustments: | ||||||||||||||||||||||
| Amortization of intangible assets | 9 | 2,323 | 2,517 | 194 | 2,323 | 2,336 | ||||||||||||||||
| Depreciation of equipment | 8 | 349 | 221 | 32 | 189 | 59 | ||||||||||||||||
| Impairment loss related to intangible assets | — | — | — | — | 339 | |||||||||||||||||
| Stock-based compensation | 16 | 929 | 674 | 86 | 588 | 309 | ||||||||||||||||
| Net financial expenses | 15 | 1,808 | 166 | 51 | 115 | (3,295 | ) | |||||||||||||||
| Realized foreign exchange gain (loss) | (7 | ) | 48 | (12 | ) | 60 | 36 | |||||||||||||||
| Deferred income tax recovery | — | (129 | ) | — | (129 | ) | — | |||||||||||||||
| Total adjustments | (16,102 | ) | (7,750 | ) | (418 | ) | (7,332 | ) | (6,533 | ) | ||||||||||||
| Changes in working capital items | 18 | 3,583 | 792 | (328 | ) | 1,120 | (41 | ) | ||||||||||||||
| Net cash used in operating activities | (12,519 | ) | (6,958 | ) | (746 | ) | (6,212 | ) | (6,574 | ) | ||||||||||||
| Cash flows from (used in) investing activities: | ||||||||||||||||||||||
| Interest received | 70 | 150 | 4 | 146 | 114 | |||||||||||||||||
| Acquisition of intangible assets | — | — | — | — | (92 | ) | ||||||||||||||||
| Acquisition of equipment | 8, 18 | (455 | ) | (2,527 | ) | (24 | ) | (2,503 | ) | (276 | ) | |||||||||||
| Acquisition of short-term investments | — | (12,765 | ) | — | (12,765 | ) | (11,954 | ) | ||||||||||||||
| Acquisition of marketable securities | (26 | ) | — | — | — | — | ||||||||||||||||
| Maturity of short-term investments | — | 22,030 | — | 22,030 | 20,437 | |||||||||||||||||
| Net cash (used in) investing activities | (411 | ) | 6,888 | (20 | ) | 6,908 | 8,229 | |||||||||||||||
| Cash flows from (used in) financing activities: | ||||||||||||||||||||||
| Net proceeds from public offering | 13(b)(c) | 11,065 | 5,010 | (34 | ) | 5,044 | — | |||||||||||||||
| Net proceeds from private placement | 12, 13(c) | (40 | ) | 1,872 | (10 | ) | 1,882 | — | ||||||||||||||
| Proceeds from exercise of warrants | 384 | — | — | — | — | |||||||||||||||||
| Share issue costs | — | — | — | — | (1 | ) | ||||||||||||||||
| Interest paid | (3 | ) | (18 | ) | — | (18 | ) | (2 | ) | |||||||||||||
| Net cash from (used in) financing activities | 11,406 | 6,864 | (44 | ) | 6,908 | (3 | ) | |||||||||||||||
| Foreign exchange (loss) gain on cash and cash equivalents held in foreign currencies | (25 | ) | (49 | ) | 9 | (58 | ) | 64 | ||||||||||||||
| Net increase (decrease) in cash and cash equivalents | (1,549 | ) | 6,745 | (801 | ) | 7,546 | 1,716 | |||||||||||||||
| Cash and cash equivalents, beginning of period | 9,772 | 3,027 | 10,573 | 3,027 | 1,311 | |||||||||||||||||
| Cash and cash equivalents, end of period | 8,223 | 9,772 | 9,772 | 10,573 | 3,027 | |||||||||||||||||
| Cash and cash equivalents is comprised of: | ||||||||||||||||||||||
| Cash | 1,583 | 6,778 | 6,778 | 7,584 | 3,027 | |||||||||||||||||
| Cash equivalents | 6,640 | 2,994 | 2,994 | 2,989 | — |
See accompanying notes to financial statements.
| F-10 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 1. | Reporting entity |
Acasti Pharma Inc. (Acasti or the Corporation) is incorporated under the Business Corporations Act (Québec) (formerly Part 1A of the Companies Act (Québec)). The Corporation is domiciled in Canada and its registered office is located at 545, Promenade du Centropolis, Laval, Québec, H7T 0A3. Neptune Technologies (Neptune) owns approximately 19.8% of the issued and outstanding Class A shares (Common Shares) of the Corporation following the US Public financing of December 27, 2017 (see note 6 and 13). Prior to the US public financing, Neptune owned approximately 34.0% of the Common Shares and was previously the parent company of Acasti.
Pursuant to a license agreement entered into with Neptune in August 2008, as amended, Acasti has been granted an exclusive worldwide license to use until its related patents expire, Neptune’s intellectual property to develop, clinically study and market new pharmaceutical and medical food products to treat human cardiovascular conditions. Neptune’s intellectual property is related to the extraction of ingredients from marine biomasses, such as krill. The eventual products are aimed at applications in the prescription drug, over-the-counter medicine and medical foods markets. In December 2012, the Corporation entered into a prepayment agreement with Neptune pursuant to which the Corporation exercised its option under the License Agreement to pay in advance all of the future royalties payable under the license which was exercised in fiscal 2014. As a result of the royalty prepayment, Acasti is no longer required to pay any royalties to Neptune under the License Agreement during its term for the use of the intellectual property under license. The license allows Acasti to exploit the intellectual property rights in order to develop novel active pharmaceutical ingredients (“APIs”) into commercial products for the prescription drugs and the medical food markets. On August 8, 2017, Neptune announced the sale of its krill oil inventory and intellectual property to Aker BioMarine Antarctic AS (Aker). Aker then licensed the intellectual property back to Neptune, leaving the License Agreement between Acasti and Neptune in place and unchanged. The license Agreement allows Acasti the “freedom to operate” for CaPre, which is currently the Corporation’s only prescription drug candidate in development. There are diligence obligations with respect to the Corporation’s use of licensed technology in relation to the development and commercialization of Acasti’s product candidate. Upon the expiry of the last-to-expire licensed Neptune patents in 2022, and the concurrent expiry of Acasti’s License Agreement with Neptune and Aker, the Corporation believes that CaPre will be fully covered under its own issued and pending patents, and after the Neptune patent expiry that Acasti will not require any license from Neptune or any other third party to support the commercialization of CaPre.
The Corporation is subject to a number of risks associated with the conduct of its clinical program and its results, the establishment of strategic alliances and the development of new pharmaceutical products and their marketing. The Corporation also knows that its current product in development requires approval from the U.S Food and Drug Administration and equivalent regulatory organizations in other countries before their sale can be authorized. The Corporation has incurred significant operating losses and negative cash flows from operations since inception. To date, the Corporation has financed its operations through the public offering and private placement of Common Shares, units consisting of Common Shares and warrants and convertible debt, the proceeds from research grants and research tax credits, and the exercises of warrants, rights and options. To achieve the objectives of its business plan, Acasti plans to raise the necessary funds through additional securities offerings and the establishment of strategic alliances as well as additional research grants and research tax credits. The ability of the Corporation to complete the needed financing andultimately achieve profitable operations is dependent on a number of factors outside of the Corporation’s control.
| 2. | Basis of preparation |
| (a) | Statement of compliance: |
These financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”). Beginning in fiscal 2017, the Corporation’s fiscal year end is on March 31. Fiscal 2017 is a transition year, and includes thirteen months of operations, beginning on March 1, 2016 and ending on March 31, 2017. As a result, for comparative purposes the above financial statements and corresponding notes to financial statements include two unaudited periods: the one-month period ended March 31, 2017 and the twelve-month period ended February 28, 2017. The Canadian Securities regulator permits, in the transition year, the presentation of a thirteen-month period for the financial year ended March 31, 2017.
The financial statements were approved by the Board of Directors on June 27, 2018.
| (b) | Basis of measurement: |
The financial statements have been prepared on the historical cost basis, except for:
| · | Stock-based compensation which is measured pursuant to IFRS 2, Share-based payments (Note 3(e) (ii)); and, |
| F-11 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| · | Derivative warrant liabilities measured at fair value on a recurring basis (Note 11). |
| 2. | Basis of preparation (continued): |
| (c) | Going concern uncertainty: |
The Corporation has incurred operating losses and negative cash flows from operations since inception. The Corporation’s current assets of $9.5 million as at March 31, 2018 include cash and cash equivalents totalling $8.2 million, mainly generated by the net proceeds from the Public Offering completed on December 27, 2017. The Corporation’s current liabilities total $6.7 million at March 31, 2018 and are comprised primarily of amounts due to or accrued for creditors. Since the Corporation’s March 31, 2018 year end, the current assets have been increased by approximately $10.0 million from the net proceeds, of a public financing completed in early May 2018 including the exercise of the over allotment option (note 24 – subsequent event). However, in spite of this incremental financing, these current assets are projected to be significantly less than what will be needed to support the current liabilities as at this date when combined with the projected level of expenses for the next twelve months, including the continued advancement of the TRILOGY Phase 3 clinical study program for its drug candidate, CaPre. Additional funds will also be needed for the expected expenses for the total CaPre Phase 3 research and development phase beyond the next twelve months, including the potential regulatory (NDA) submission. The Corporation also expects to incur increased general and administrative expenses as a result of a planned increase in business development and commercialization planning expenses, and a reduction of its shared services agreement with Neptune, with those added expenses having begun during the year ended March 31, 2018. In addition to the recently raised additional funds, the Corporation is working towards development of strategic partner relationships and plans to raise additional funds in the future, but there can be no assurance as to when or whether Acasti will complete any additional financing or strategic collaborations. In particular, raising financing is subject to market conditions and is not within the Corporation’s control. If the Corporation does not raise additional funds, find one or more strategic partners, it may not be able to realize its assets and discharge its liabilities in the normal course of business. As a result, there exists a material uncertainty that casts substantial doubt about the Corporation’s ability to continue as a going concern and, therefore, realize its assets and discharge its liabilities in the normal course of business. The Corporation currently has no other arranged sources of financing.
The financial statements have been prepared on a going concern basis, which assumes the Corporation will continue its operations in the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the ordinary course of business. These financial statements do not include any adjustments to the carrying values and classification of assets and liabilities and reported expenses that may be necessary if the going concern basis was not appropriate for these financial statements. If the Corporation was unable to continue as a going concern, material write-downs to the carrying values of the Corporation’s assets, including the intangible asset, could be required.
| (d) | Functional and presentation currency: |
These financial statements are presented in Canadian dollars, which is the Corporation’s functional currency.
| (e) | Use of estimates and judgments: |
The preparation of the financial statements in conformity with IFRS requires management to make judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets, liabilities, income and expenses. Actual results may differ from these estimates.
Estimates are based on management’s best knowledge of current events and actions that the Corporation may undertake in the future. Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected.
Critical judgments in applying accounting policies that have the most significant effect on the amounts recognized in the financial statements include the following:
| · | Identification of triggering events indicating that the intangible assets might be impaired. |
| · | The use of the going concern basis of preparation of the financial statements. At the end of each reporting period, management assesses the basis of preparation of the financial statements (Note 2(c)). |
| F-12 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 2. | Basis of preparation (continued): |
| (e) | Use of estimates and judgments (continued): |
Assumptions and estimation uncertainties that have a significant risk of resulting in a material adjustment within the next financial year include the following:
| · | Determination of the recoverable amount of the Corporation’s cash generating unit (“CGU”). |
| · | Measurement of derivative warrant liabilities (note 11) and stock-based compensation (note 16). |
Also, management uses judgment to determine which research and development (“R&D”) expenses qualify for R&D tax credits and in what amounts. The Corporation recognizes the tax credits once it has reasonable assurance that they will be realized. Recorded tax credits are subject to review and approval by tax authorities and therefore, could be different from the amounts recorded.
| 3. | Significant accounting policies: |
The accounting policies set out below have been applied consistently to all periods presented in these financial statements.
| (a) | Financial instruments: |
A financial instrument is any contract that gives rise to a financial asset of one party and a financial liability or equity instrument of another party.
| (i) | Non-derivative financial assets: |
The Corporation has the following non-derivative financial assets: cash, cash equivalents, marketable securities and receivables. The Corporation determines the classification of its financial assets at initial recognition. The subsequent measurement of financial assets depends on their classification.
Financial assets and liabilities are offset and the net amount presented in the statements of financial position when, and only when, the Corporation has a legal right to offset the amounts and intends either to settle on a net basis or to realize the asset and settle the liability simultaneously.
Loans and receivables
The classification “loans and receivables” comprises financial assets with fixed or determinable payments that are not quoted in an active market. Such assets are recognized initially at fair value plus any directly attributable transaction costs. Subsequent to initial recognition, loans and receivables are measured at amortized cost using the effective interest method, less any impairment losses.
Cash, cash equivalents, marketable securities and receivables with maturities of less than one year are classified as loans and receivables.
Cash and cash equivalents comprise cash balances and highly liquid investments purchased three months or less from maturity.
| (ii) | Non-derivative financial liabilities: |
The Corporation has the following non-derivative financial liabilities: trade and other payables, and unsecured convertible debentures. Such financial liabilities are recognized initially at fair value plus any directly attributable transaction costs. Subsequent to initial recognition, these financial liabilities are measured at amortized cost using the effective interest method.
The Corporation derecognizes a financial liability when its contractual obligations are discharged, cancelled or expire.
| F-13 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (a) | Financial instruments (continued): |
| (iii) | Compound financial instruments: |
Compound financial instruments are instruments that can be converted to share capital at the option of the holder, and the number of shares to be issued is fixed.
The unsecured convertible debentures are compound instruments and have been separated into liability and equity components. The liability component is recognized initially at the fair value of a similar liability that does not have an equity conversion option. The equity component is recognized initially as the difference between the fair value of the compound financial instrument as a whole and the fair value of the liability component. Any directly attributable transaction costs are allocated to the liability and equity components in proportion to their initial carrying amounts. Subsequent to initial recognition, the liability component of a compound financial instrument is measured at amortized cost using the effective interest method. The equity component of a compound financial instrument is not remeasured subsequent to initial recognition.
| (iv) | Share capital: |
Common Shares
Class A Common Shares are classified as equity. Incremental costs directly attributable to the issue of Common Shares and share options are recognized as a deduction from share capital, net of any tax effects.
| (v) | Derivative financial instruments: |
The Corporation has issued liability-classified derivatives over its own equity. Derivatives are recognized initially at fair value; attributable transaction costs are recognized in profit and loss as incurred. Subsequent to initial recognition, derivatives are measured at fair value, and all changes in their fair value are recognized immediately in profit or loss.
| (vi) | Other equity instruments: |
Warrants, options and rights over the Corporation’s equity issued outside of share-based payment transactions that do not meet the definition of a liability instrument are recognized in equity.
| (b) | Equipment: |
| (i) | Recognition and measurement: |
Equipment is measured at cost less accumulated depreciation and accumulated impairment losses, if any.
Cost includes expenditures that are directly attributable to the acquisition of the asset, including all costs incurred in bringing the asset to its present location and condition.
Purchased software that is integral to the functionality of the related equipment is capitalized as part of that equipment.
Gains and losses on disposal of equipment are determined by comparing the proceeds from disposal with the carrying amount of equipment, and are recognized net within ''other income or expenses'' in profit or loss.
| (ii) | Subsequent costs: |
The cost of replacing a part of an equipment is recognized in the carrying amount of the item if it is probable that the future economic benefits embodied within the part will flow to the Corporation, and its cost can be measured reliably. The carrying amount of the replaced part is derecognized. The costs of the day-to-day servicing of equipment are recognized in profit or loss as incurred.
| F-14 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (b) | Equipment (continued): |
| (iii) | Depreciation: |
Depreciation is recognized in profit or loss on either a straight-line basis or a declining basis over the estimated useful lives of each part of an item of equipment, since this most closely reflects the expected pattern of consumption of the future economic benefits embodied in the asset. Items of equipment are depreciated from the date that they are available for use or, in respect of assets not yet in service, from the date they are ready for their intended use.
The estimated useful lives and rates for the current and comparative periods are as follows:
| Assets | Method | Period/Rate | ||
| Furniture and office equipment | Declining balance | 20% | to | 30% |
| Computer equipment | Declining balance | 30% | ||
| Laboratory equipment | Declining balance | 30% | ||
| Production equipment (in years) | Straight-line | 10 | ||
Depreciation methods, useful lives and residual values are reviewed at each financial year-end and adjusted prospectively if appropriate.
| (c) | Intangible assets: |
| (i) | Research and development: |
Expenditure on research activities, undertaken with the prospect of gaining new scientific or technical knowledge and understanding, is recognized in profit or loss as incurred.
Development activities involve a plan or design for the production of new or substantially improved products and processes. Development expenditure is capitalized only if development costs can be measured reliably, the product or process is technically and commercially feasible, future economic benefits are probable, and the Corporation intends to and has sufficient resources to complete development and to use or sell the asset. The expenditure capitalized includes the cost of materials, direct labour, overhead costs that are directly attributable to preparing the asset for its intended use, and borrowing costs on qualifying assets. Other development expenditures are recognized in profit or loss as incurred.
Capitalized development expenditure is measured at cost less accumulated amortization and accumulated impairment losses. As of the reporting periods presented, the Corporation has not capitalized any development expenditure.
| (ii) | Other intangible assets: |
Patent costs
Patents for technologies that are no longer in the research phase are recorded at cost. Patent costs include legal fees to obtain patents and patent application fees. When the technology is still in the research and development phase, those costs are expensed as incurred.
Licenses
Licenses that are acquired by the Corporation and have finite useful lives are measured at cost less accumulated amortization and accumulated impairment losses.
| F-15 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (c) | Intangible assets (continued): |
| (iii) | Subsequent expenditure: |
Subsequent expenditure is capitalized only when it increases the future economic benefits embodied in the specific asset to which it relates. All other expenditures, including expenditure on internally generated goodwill and brands, are recognized in profit or loss as incurred.
| (iv) | Amortization: |
Amortization is calculated over the cost of the intangible asset less its residual value.
Amortization is recognized in profit or loss on a straight-line basis over the estimated useful lives of intangible assets from the date that they are available for use, since this most closely reflects the expected pattern of consumption of the future economic benefits embodied in the asset. The estimated useful lives for the current and comparative periods are as follows:
| Assets | Period (in years) | ||
| Patents | 20 | ||
| License | 8 | to | 14 |
| (d) | Impairment: |
| (i) | Financial assets: |
A financial asset not carried at fair value through profit or loss is assessed at each reporting date to determine whether there is objective evidence that it is impaired. A financial asset is impaired if objective evidence, such as default or delinquency by a debtor, indicates that a loss event has occurred after the initial recognition of the asset, and that the loss event had a negative effect on the estimated future cash flows of that asset that can be estimated reliably.
An impairment loss in respect of a financial asset measured at amortized cost is calculated as the difference between its carrying amount and the present value of the estimated future cash flows discounted at the asset’s original effective interest rate. Losses are recognized in profit or loss and reflected in an allowance account against the financial asset. When a subsequent event causes the amount of impairment loss to decrease, the decrease in impairment loss is reversed through profit or loss.
| (ii) | Non-financial assets: |
The carrying amounts of the Corporation’s non-financial assets are reviewed at each reporting date to determine whether there is any indication of impairment. If any such indication exists, then the asset’s recoverable amount is estimated.
The recoverable amount of an asset or cash-generating unit is the greater of its value in use and its fair value less costs to sell. In assessing value in use, the estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the risks specific to the asset. For the purpose of impairment testing, assets that cannot be tested individually are grouped together into the smallest group of assets that generates cash inflows from continuing use that are largely independent of the cash inflows of other assets or groups of assets (the “cash-generating unit, or “CGU”).
| F-16 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (d) | Impairment (continued): |
| (ii) | Non-financial assets (continued): |
The Corporation’s corporate assets do not generate separate cash inflows. If there is an indication that a corporate asset may be impaired, then the recoverable amount is determined for the CGU to which the corporate asset belongs.
An impairment loss is recognized if the carrying amount of an asset or its CGU exceeds its estimated recoverable amount. Impairment losses are recognized in profit or loss.
Impairment losses recognized in prior years are assessed at each reporting date for any indications that the loss has decreased or no longer exists. An impairment loss is reversed if there has been a change in the estimates used to determine the recoverable amount. An impairment loss is reversed only to the extent that the asset’s carrying amount does not exceed the carrying amount that would have been determined, net of depreciation or amortization, if no impairment loss had been recognized.
| (e) | Employee benefits: |
| (i) | Short-term employee benefits: |
Short-term employee benefit obligations are measured on an undiscounted basis and are expensed as the related service is provided.
A liability is recognized for the amount expected to be paid under short-term cash bonus or profit-sharing plans if the Corporation has a present legal or constructive obligation to pay this amount as a result of past service provided by the employee, and the obligation can be estimated reliably.
| (ii) | Share-based payment transactions: |
The grant date fair value of share-based payment awards granted to employees is recognized as an employee expense, with a corresponding increase in contributed surplus, over the period that the employees unconditionally become entitled to the awards. The grant date fair value takes into consideration market performance conditions when applicable. The amount recognized as an expense is adjusted to reflect the number of awards for which the related service and non-market vesting conditions are expected to be met, such that the amount ultimately recognized as an expense is based on the number of awards that do meet the related service and non-market performance conditions at the vesting date.
Share-based payment arrangements in which the Corporation receives goods or services as consideration for its own equity instruments are accounted for as equity-settled share-based payment transactions, regardless of how the equity instruments are obtained by the Corporation.
| (iii) | Termination benefits: |
Termination benefits are recognized as an expense when the Corporation is committed demonstrably, without realistic possibility of withdrawal, to a formal detailed plan to either terminate employment before the normal retirement date, or to provide termination benefits as a result of an offer made to encourage voluntary redundancy. Termination benefits for voluntary redundancies are recognized as an expense if the Corporation has made an offer of voluntary redundancy, it is probable that the offer will be accepted, and the number of acceptances can be estimated reliably. If benefits are payable more than 12 months after the reporting year, then they are discounted to their present value.
| F-17 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (f) | Provisions: |
A provision is recognized if, as a result of a past event, the Corporation has a present legal or constructive obligation that can be estimated reliably, and it is probable that an outflow of economic benefits will be required to settle the obligation. Provisions are determined by discounting the expected future cash flows at a pre-tax rate that reflects current market assessments of the time value of money and the risks specific to the liability. The unwinding of the discount is recognized as finance cost.
| (i) | Onerous contracts: |
A provision for onerous contracts is recognized when the expected benefits to be derived by the Corporation from a contract are lower than the unavoidable cost of meeting its obligations under the contract. The provision is measured at the present value of the lower of the expected cost of terminating the contract and the expected net cost of continuing with the contract. Before a provision is established, the Corporation recognizes any impairment loss on the assets associated with that contract.
| (ii) | Contingent liability: |
A contingent liability is a possible obligation that arises from past events and of which the existence will be confirmed only by the occurrence or non-occurrence of one or more uncertain future events not within the control of the Corporation; or a present obligation that arises from past events (and therefore exists), but is not recognized because it is not probable that a transfer or use of assets, provision of services or any other transfer of economic benefits will be required to settle the obligation; or the amount of the obligation cannot be estimated reliably.
| (g) | Government grants: |
Government grants are recorded as a reduction of the related expense or cost of the asset acquired. Government grants are recognized when there is reasonable assurance that the Corporation has met the requirements of the approved grant program and there is reasonable assurance that the grant will be received.
Grants that compensate the Corporation for expenses incurred are recognized in profit or loss in reduction thereof on a systematic basis in the same years in which the expenses are recognized. Grants that compensate the Corporation for the cost of an asset are recognized in profit or loss on a systematic basis over the useful life of the asset.
| (h) | Lease payments: |
Payments made under operating leases are recognized in profit or loss on a straight-line basis over the term of the lease. Lease incentives received are recognized as an integral part of the total lease expense, over the term of the lease.
| (i) | Foreign currency: |
Transactions in foreign currencies are translated into the functional currency at exchange rates at the dates of the transactions. Monetary assets and liabilities denominated in foreign currencies at the reporting date are translated to the functional currency at the exchange rate at that date. The foreign currency gain or loss on monetary items is the difference between amortized cost in the functional currency at the beginning of the period, adjusted for effective interest and payments during the period, and the amortized cost in foreign currency translated at the exchange rate at the end of the reporting period. Foreign currency differences arising on translation are recognized in profit or loss.
| F-18 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (j) | Finance income and finance expense: |
Finance income comprises interest income on funds invested. Interest income is recognized as it accrues in profit or loss, using the effective interest method.
Finance costs comprise interest expense, accretion on borrowings, unwinding of the discount on provisions, impairment losses recognized on financial assets and transaction costs for issuance of derivative warrant liabilities. Borrowing costs that are not directly attributable to the acquisition, construction or production of a qualifying asset are recognized in profit or loss using the effective interest method.
Foreign currency gains and losses are reported on a net basis.
The Corporation recognizes interest income as a component of investing activities and interest expense as a component of financing activities in the statements of cash flows.
| (k) | Income tax: |
Income tax expense comprises current and deferred taxes. Current and deferred taxes are recognized in profit or loss except to the extent that they relate to items recognized directly in equity or in other comprehensive income.
Current tax is the expected tax payable or receivable on the taxable income or loss for the year, using tax rates enacted at the reporting date, and any adjustment to tax payable in respect of previous years.
Deferred tax is recognized in respect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for taxation purposes. Deferred tax is not recognized for temporary differences arising from the initial recognition of assets or liabilities in a transaction that is not a business combination and that affects neither accounting nor taxable profit or loss. Deferred tax is measured at the tax rates, enacted or substantively enacted, that are expected to be applied to temporary differences when they reverse, based on the laws that have been enacted or substantively enacted by the reporting date. Deferred tax assets and liabilities are offset if there is a legally enforceable right to offset current tax liabilities and assets, and they relate to income taxes levied by the same tax authority on the same taxable entity, or on different tax entities, but they intend to settle current tax liabilities and assets on a net basis or their tax assets and liabilities will be realized simultaneously. A deferred tax asset is recognized for unused tax losses, tax credits and deductible temporary differences, to the extent that it is probable that future taxable profits will be available against which they can be utilized. Deferred tax assets are reviewed at each reporting date and are reduced to the extent that it is no longer probable that the related tax benefit will be realized.
| (l) | Earnings per share: |
The Corporation presents basic and diluted earnings per share (“EPS”) data for its Class A shares (or “Common Shares”). Basic EPS is calculated by dividing the profit or loss attributable to the holders of Class A shares (Common Shares) of the Corporation by the weighted average number of Common Shares outstanding during the year, adjusted for own shares held. Diluted EPS is determined by adjusting the profit or loss attributable to the holders of Class A shares (Common Shares) and the weighted average number of Class A shares (Common Shares) outstanding adjusted for the effects of all dilutive potential Common Shares, which comprise warrants, rights and share options granted to employees.
| (m) | Segment reporting: |
An operating segment is a component of the Corporation that engages in business activities from which it may earn revenues and incur expenses. The Corporation has one reportable operating segment: the development and commercialization of pharmaceutical applications of its licensed rights for cardiovascular diseases. The majority of the Corporation’s assets are located in Canada, while one major production unit, with a carrying value of $2,077 (March 31, 2017-$2,394), is located in France.
| F-19 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 3. | Significant accounting policies (continued): |
| (n) | Change in accounting policy: |
Future accounting change:
The following new standards, and amendments to standards and interpretations, are not yet effective for the period ended March 31, 2018, and have not been applied in preparing these financial statements.
New standards and interpretations not yet adopted:
(i) Financial instruments:
On July 24, 2014, the International Accounting Standards Board (IASB) issued the final version of IFRS 9, Financial Instruments, replacing IAS 39, Financial Instruments: Recognition and Measurement. IFRS 9 introduces a revised approach for the classification of financial assets based on how an entity manages financial assets and the characteristics of the contractual cash flows of the financial assets replacing the multiple rules in IAS 39. Most of the requirements in IAS 39 for classification and measurement of financial liabilities have been carried forward in IFRS 9. IFRS 9 also introduces a new hedge accounting model that is more closely aligned with risk-management activities and a new expected credit loss model for calculating impairment on financial assets replacing the incurred loss model in IAS 39.
IFRS 9 is effective for annual periods beginning on or after January 1, 2018, with earlier adoption permitted. The Corporation intends to adopt IFRS 9 in its financial statements for the annual period beginning on April 1, 2018.
The Company’s preliminary analysis has not identified any significant differences in respect to the classification and measurement of financial instruments and continues to evaluate the impact of the new standard on its financial statements. The Corporation does not apply hedge accounting.
(ii) Amendments to IFRS 2 – Classification and Measurement of Share-Based Payment Transactions:
On June 20, 2016, the IASB issued amendments to IFRS 2, Share-Based Payment, clarifying how to account for certain types of share-based payment transactions. The amendments apply for annual periods beginning on or after January 1, 2018. Earlier application is permitted. As a practical simplification, the amendments can be applied prospectively. Retrospective, or early application is permitted if information is available without the use of hindsight. The amendments provide requirements on the accounting for: the effects of vesting and non-vesting conditions on the measurement of cash-settled share-based payments; share-based payment transactions with a net settlement feature for withholding tax obligations; and a modification to the terms and conditions of a share-based payment that changes the classification of the transaction from cash-settled to equity-settled. The Corporation intends to adopt the amendments to IFRS 2 in its financial statements for the annual period beginning on April 1, 2018. The Corporation has not yet assessed the impact of adoption of the amendments of IFRS 2.
| 4. | Receivables: |
| March 31, 2018 | March 31, 2017 | February 28, 2017 | ||||||||||||
| Notes | $ | $ | (Unaudited) $ |
|||||||||||
| Sales tax receivables | 470 | 89 | 83 | |||||||||||
| Government assistance and tax credits receivable | 7 | 282 | 115 | 81 | ||||||||||
| Other receivables | 7 | 2 | 2 | |||||||||||
| Total receivables | 759 | 206 | 166 |
| 5. | Other Assets |
During the year, the Corporation purchased a reserve of krill oil amounting to $970 to be used in production. The krill oil is expensed as it is used in the R&D production processes for NKPL66 manufacturing. $259 of krill oil from the reserve was used for the manufacturing of CaPre capsules as at March 31, 2018, as well as a credit of $52 was received for damaged drums, leaving a balance of $659 of which an amount of $104 is estimated to be used in the next twelve-month period.
| F-20 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 6. | Related parties: |
| (a) | Administrative and research and development expenses: |
The Corporation intends to continue to rely on the support of Neptune for a portion of its general and administrative needs; however, the continuance of this support is outside of the Corporation’s control. The Corporation was charged by Neptune for the purchase of research supplies and for certain costs incurred by Neptune for the benefit of the Corporation, as follows:
| Thirteen-months ended |
Month ended | Twelve-months ended |
||||||||||||||||||
| March 31, 2018 | March 31, 2017 | March 31, 2017 | February 28, 2017 | February 29, 2016 | ||||||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Research and development expenses | ||||||||||||||||||||
| Supplies and incremental costs | 7 | - | - | - | 5 | |||||||||||||||
| Shared service agreement | 20 | 60 | 1 | 59 | 366 | |||||||||||||||
| Total | 27 | 60 | 1 | 59 | 371 | |||||||||||||||
| General and administrative expenses | ||||||||||||||||||||
| Supplies and incremental costs | 239 | 293 | 16 | 277 | 299 | |||||||||||||||
| Shared service agreement | 121 | 325 | 25 | 300 | 491 | |||||||||||||||
| Total | 360 | 618 | 41 | 577 | 790 | |||||||||||||||
| Total related parties expenses | 387 | 678 | 42 | 636 | 1,161 |
Where Neptune incurs specific incremental costs for the benefit of the Corporation, it charges those amounts directly. Neptune provides Acasti with the services of personnel for certain administrative work as part of a shared service agreement. The employees’ salaries and benefits are charged proportionally to the time allocation agreed upon within the shared service agreement. For the year ended March 31, 2018 laboratory support, the corporate affairs and the public company reporting services previously provided by Neptune as part of the shared service agreement were discontinued. The Corporation is now incurring incremental costs and expects to do so in the future, partially offset by reduced shared service fees. The account payable to Neptune amounted to $44 at March 31, 2018, $12 at March 31, 2017, and $15 at February 29, 2016, is non-interest bearing and has no specified maturity date. These charges do not represent all charges incurred by Neptune that may have benefited the Corporation. Also, these charges do not necessarily represent the cost that the Corporation would otherwise need to incur, should it not receive these services or benefits through the shared resources of Neptune.
Historically, Neptune has provided the Corporation with the krill oil needed to produce CaPre for Acasti’s clinical programs, including all of the krill oil projected as needed for its Phase 3 clinical study program. However, Neptune discontinued its krill oil production and sold its krill oil inventory to Aker on August 7, 2017. During the period, Acasti purchased a reserve of krill oil from Aker that will be used in the production of CaPre capsules for its Phase 3 clinical trials (see also note 5). The Corporation is currently evaluating alternative suppliers of krill oil. At March 31, 2018, a reserve of krill oil was still stored at Neptune’s facility.
| (b) | Interest revenue: |
On January 7, 2016 Neptune announced the acquisition of Biodroga Nutraceuticals Inc. As part of this transaction, the Corporation pledged an amount of $2 million (“Committed Funds”) to partly guarantee the financing for the said transaction (“Pledge Agreement”). Neptune had agreed to pay Acasti an annual fee on the Committed Funds outstanding at an annual rate of 9% during the first six months and 11% for the remaining term of the Pledge Agreement. On September 20, 2016, Neptune fully released the pledged amount. The Corporation recognized interest revenue of nil for the year ended March 31, 2018 and, $89 for the thirteen-month period ended March 31, 2017, nil (unaudited) for the month ended March 31, 2017, and $89 (unaudited) for the twelve-month period ended February 28, 2017 and $27 for the year ended February 29, 2016.
| F-21 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 6. | Related parties (continued): |
| (c) | Key management personnel compensation: |
The key management personnel are the officers of the Corporation and the members of the Board of Directors of the Corporation. They control in the aggregate less than 1% of the voting shares of the Corporation (2% in 2017 and 1% in 2016).
Key management personnel compensation includes the following for the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, twelve-month period ended February 28, 2017, and year ended February 29, 2016.
| Thirteen- months ended |
Month ended | Twelve- months ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 |
February 28, 2017 |
February 29, 2016 |
||||||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Compensation |
1,754 |
1,510 |
146 |
1,364 |
688 |
|||||||||||||||
| Severance | - | - | - | - | 103 | |||||||||||||||
| Share-based compensation costs | 826 | 619 | 78 | 541 | 120 | |||||||||||||||
| Total key management personnel compensation | 2,580 | 2,129 | 224 | 1,905 | 911 |
| 7. | Government assistance: |
| Thirteen- months ended |
Month ended | Twelve- months ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 | February 28, 2017 | February 29, 2016 | ||||||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Investment tax credit | 409 | 103 | 8 | 95 | 169 | |||||||||||||||
| Government grant | - | 227 | 37 | 190 | 180 | |||||||||||||||
| Total government assistance | 409 | 330 | 45 | 285 | 349 |
Government assistance is comprised of a government grant from the federal government and research and development investment tax credits receivable from the provincial government which relate to qualifiable research and development expenditures under the applicable tax laws. The amounts recorded as receivables are subject to a government tax audit and the final amounts received may differ from those recorded.
| F-22 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 7. | Government assistance (continued): |
Unrecognized federal tax credits may be used to reduce future income tax and expire as follows:
| $ | ||||
| 2029 | 11 | |||
| 2030 | 30 | |||
| 2031 | 45 | |||
| 2032 | 431 | |||
| 2033 | 441 | |||
| 2034 | 436 | |||
| 2035 | 519 | |||
| 2036 | 286 | |||
| 2037 | 315 | |||
| 2038 | 345 | |||
| 2,859 |
| F-23 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 8. | Equipment: |
| Furniture and office equipment |
Computer equipment |
Laboratory equipment |
Production equipment |
Total | ||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Cost: | ||||||||||||||||||||
| Balance at February 28, 2015 | 59 | 3 | 60 | — | 122 | |||||||||||||||
| Additions | — | — | 276 | — | 276 | |||||||||||||||
| Balance at February 29, 2016 | 59 | 3 | 336 | — | 398 | |||||||||||||||
| Additions for the twelve-month period (Unaudited) | — | 8 | 186 | 2,484 | 2,678 | |||||||||||||||
| Balance at February 28, 2017 (Unaudited) | 59 | 11 | 522 | 2,484 | 3,076 | |||||||||||||||
| Additions for the one-month period (Unaudited) | — | — | — | 43 | 43 | |||||||||||||||
| Additions for the thirteen-month period | — | 8 | 186 | 2,527 | 2,721 | |||||||||||||||
| Balance at March 31, 2017 | 59 | 11 | 522 | 2,527 | 3,119 | |||||||||||||||
| Additions | 4 | 6 | 192 | 181 | 383 | |||||||||||||||
| Balance at March 31, 2018 | 63 | 17 | 714 | 2,708 | 3,502 | |||||||||||||||
| Accumulated depreciation: | ||||||||||||||||||||
| Balance at February 28, 2015 | 49 | 3 | — | — | 52 | |||||||||||||||
| Depreciation for the year | 3 | — | 56 | — | 59 | |||||||||||||||
| Balance at February 29, 2016 | 52 | 3 | 56 | — | 111 | |||||||||||||||
| Depreciation for the twelve-month period (Unaudited) | 7 | 1 | 129 | 52 | 189 | |||||||||||||||
| Balance at February 28, 2017 (Unaudited) | 59 | 4 | 185 | 52 | 300 | |||||||||||||||
| Depreciation for the one-month period (Unaudited) | — | — | 11 | 21 | 32 | |||||||||||||||
| Depreciation for thirteen-month period | 7 | 1 | 140 | 73 | 221 | |||||||||||||||
| Balance at March 31, 2017 | 59 | 4 | 196 | 73 | 332 | |||||||||||||||
| Depreciation | — | 3 | 107 | 239 | 349 | |||||||||||||||
| Balance at March 31, 2018 | 59 | 7 | 303 | 312 | 681 | |||||||||||||||
| Net carrying amounts: | ||||||||||||||||||||
| February 28, 2017 (Unaudited) | — | 7 | 337 | 2,432 | 2,776 | |||||||||||||||
| March 31, 2017 | — | 7 | 326 | 2,454 | 2,787 | |||||||||||||||
| March 31, 2018 | 4 | 10 | 411 | 2,396 | 2,821 |
Depreciation expense for the period end March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017 and twelve-month period ended February 28, 2017 has been recorded in “research and development expenses” in the statements of earnings and comprehensive loss.
During the year a reclassification of $94 cost related to tooling for the thirteen-month period ended March 31, 2017 was made between production equipment and prepaid assets. No depreciation was taken in relation to these amounts.
| F-24 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 9. | Intangible assets : |
| Patents | License | Total | ||||||||||
| $ | $ | $ | ||||||||||
| Cost: | ||||||||||||
| Balance at February 28, 2015 | 278 | 24,330 | 24,608 | |||||||||
| Additions | 84 | — | 84 | |||||||||
| Balance at February 29, 2016, February 28, 2017 (Unaudited) and March 31, 2017 | 362 | 24,330 | 24,692 | |||||||||
| Additions | — | — | — | |||||||||
| Balance at March 31, 2018 | 362 | 24,330 | 24,692 | |||||||||
| Accumulated amortization: | ||||||||||||
| Balance at February 28, 2015 | 10 | 7,102 | 7,112 | |||||||||
| Amortization for the year | 13 | 2,323 | 2,336 | |||||||||
| Impairment loss | 339 | — | 339 | |||||||||
| Balance at February 29, 2016 | 362 | 9,425 | 9,787 | |||||||||
| Amortization for the twelve-month period (Unaudited) | — | 2,323 | 2,323 | |||||||||
| Balance at February 28, 2017 (Unaudited) | 362 | 11,748 | 12,110 | |||||||||
| Amortization for the one-month period (Unaudited) | — | 194 | 194 | |||||||||
| Amortization for the thirteen-month period | — | 2,517 | 2,517 | |||||||||
| Balance at March 31, 2017 | 362 | 11,942 | 12,304 | |||||||||
| Amortization for the year | — | 2,323 | 2,323 | |||||||||
| Balance at March 31, 2018 | 362 | 14,265 | 14,627 | |||||||||
| Net carrying amounts: | ||||||||||||
| February 28, 2017 (Unaudited) | — | 12,582 | 12,582 | |||||||||
| March 31, 2017 | — | 12,388 | 12,388 | |||||||||
| March 31, 2018 | — | 10,065 | 10,065 |
Amortization expense and impairment loss for the period ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 have been recorded in “research and development expenses” in the statements of earnings and comprehensive loss.
| F-25 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 10. | Trade and other payables: |
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||
| $ | $ | $ | ||||||||||
| Trade payables | 3,420 | 259 | 534 | |||||||||
| Accrued liabilities and other payables | 2,479 | 1,354 | 1,372 | |||||||||
| Employee salaries and benefits payable | 754 | 513 | 484 | |||||||||
| Payable to Neptune | 44 | 12 | 15 | |||||||||
| Total trade and other payables | 6,697 | 2,138 | 2,405 |
The Corporation’s exposure to currency and liquidity risks related to trade and other payables is presented in Note 20.
| 11. | Derivative warrant liabilities: |
Warrants issued as part of a public offering of units composed of class A share (Common Share) and Common Share purchase warrants on both December 27, 2017 and December 3, 2013 are derivative liabilities (“Derivative warrant liabilities”) given the currency of the exercise price is different from the Corporation’s functional currency.
The derivative warrant liabilities are measured at fair value at each reporting period and the reconciliation of changes in fair value is presented in the following tables:
| Warrant liabilities issued December 27, 2017 | ||||||||||||||||
| Thirteen-month period | Month ended | Twelve-month period | ||||||||||||||
| March 31, 2018 | Ended, March 31, 2017 | March 31, 2017 (Unaudited) |
ended February 28, 2017 (Unaudited) |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Balance – beginning of period | - | - | - | - | ||||||||||||
| Issued during period (note 13b) | 5,873 | - | - | - | ||||||||||||
| Change in fair value of derivative warrant liabilities | 532 | - | - | - | ||||||||||||
| Balance – end of period | 6,405 | - | - | - | ||||||||||||
| Warrant liabilities issued December 3, 20131 | ||||||||||||||||
| Thirteen-month period | Month ended | Twelve-month period | ||||||||||||||
| March 31, 2018 | ended, March 31, 2017 | March 31, 2017 (Unaudited) |
ended February 28, 2017 (Unaudited) |
|||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Balance – beginning of period | 209 | 156 | 187 | 156 | ||||||||||||
| Change in fair value of derivative warrant liabilities | (188 | ) | 53 | 22 | 31 | |||||||||||
| Balance – end of period | 21 | 209 | 209 | 187 | ||||||||||||
(1) In order to obtain one Common Share, 10 warrants must be exercised.
| F-26 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 11. | Derivative warrant liabilities (continued): |
The fair value of the derivative warrant liabilities was estimated using the Black-Scholes option pricing model and based on the following assumptions:
| Warrant liabilities issued December 27, 2017 | ||||||||||||
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||
| Exercise price | US $1.26 | — | — | |||||||||
| Share price | US $1.02 | — | — | |||||||||
| Dividend | — | — | — | |||||||||
| Risk-free interest | 2.56 | % | — | — | ||||||||
| Estimated life (in years) | 4.75 | — | — | |||||||||
| Expected volatility | 95.16 | % | — | — | ||||||||
The fair value of the warrants issued was determined to be $0.65 per share issuable (nil as at March 31, 2017 and February 28, 2017).
| Warrant liabilities issued December 3, 20131 | ||||||||||||
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||
| Exercise price | US $1.50 | US $1.50 | US $1.50 | |||||||||
| Share price(1) | US $1.02 | US $1.36 | US $1.25 | |||||||||
| Dividend | — | — | — | |||||||||
| Risk-free interest | 2.19 | % | 1.22 | % | 1.24 | % | ||||||
| Estimated life (in years) | 0.68 | 1.68 | 1.76 | |||||||||
| Expected volatility | 133.86 | % | 108.35 | % | 107.36 | % | ||||||
(1) In order to obtain one Common Share, 10 warrants must be exercised.
The fair value of the warrants issued was determined to be $0.01 ($0.11 per share issuable as at March 31, 2017 and $0.10 (unaudited) per share issuable as at February 28, 2017).
| F-27 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 12. | Unsecured convertible debentures |
Concurrent with the Public Offering described in note 11, on February 21, 2017, the Company issued $2,000 aggregate principal amount of unsecured convertible debentures maturing February 21, 2020 and contingent warrants to acquire up to 1,052,630 Common Shares (the “Private Placement”). The principal may be prepaid, in whole or in part, at any time and from time to time, in cash, at the sole discretion of the Corporation. The debentures are convertible into Common Shares at any time by the holder at a fixed price of $1.90 per Common Share except if the Corporation pays before the maturity, all or any portion of the convertible debentures. Should the Corporation pay all or any portion of the convertible debenture before maturity, then warrants become exercisable at $1.90 per Common Share for the equivalent convertible debenture amount prepaid. The contingent warrants will be exercisable for the remaining term of the convertible debt for the same price as the conversion options. The unsecured convertible debentures were issued at a discount of 3.5% to the principal amount, for aggregate gross proceeds of $1,930.
The convertible debentures provide the Corporation an accelerated conversion right whereby the Corporation may, at any time at least four months after the date of issuance of the convertible debentures, accelerate the conversion of the debentures to Common Shares in the event that the volume weighted average price of the Corporation’s Common Shares on the TSX Venture Exchange is equal to or exceeds $2.65, subject to customary adjustment provisions, during 20 consecutive trading days.
The interest to be paid on the convertible debentures under the terms of the agreement is 8% per annum, payable on a quarterly basis in cash or Common Shares of the Corporation or a combination thereof, commencing on March 31, 2017. The decision to pay the interest due in cash or shares is at the discretion of the Corporation and the number of Common Shares to be issued will be calculated at the current market price as at the close of business on the day before the interest payment is to be made. Payment in shares shall be at a floor price of $0.10 per share, with the difference between the amount payable and the amount computed at floor price payable in cash.
The proceeds of the Private Placement were split between the liability and the equity at the time of issuance of the Private Placement. Both the conversion option and contingent warrants are considered the equity component of the Private Placement. The fair value of the liability component was determined through a discounted cash flow analysis using a discount rate of 20% that was set based on a similar debt and maturity considering the Corporation’s credit risk excluding the conversion option and contingent warrants. The amount allocated to the equity component is the residual amount after deducting the fair value of the financial liability component from the fair value of the entire compound instrument. Subsequent to initial recognition, the liability is measured at amortized cost calculated using the effective interest rate method and will accrete up to the principal balance at maturity. The interest accretion is presented as a financial expense. The equity component is not re-measured. Transaction costs were allocated to the components in proportion to their initial carrying amounts. The portion allocated to the liability was recognized as a reduction of the debt whereas the portion allocated to other equity was recognized as a reduction to other equity.
| F-28 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 12. | Unsecured convertible debentures (continued): |
The split between the liability and equity component portions of the Private Placement are summarized below:
| Liability component | Equity component | Total Private Placement |
||||||||||
| $ | $ | $ | ||||||||||
| Components at date of issue | 1,519 | 481 | 2,000 | |||||||||
| Transaction costs and debt discount | (134 | ) | (43 | ) | (177 | ) | ||||||
| Deferred income tax expense (note 18) | — | (129 | ) | (129 | ) | |||||||
| Effective interest for the twelve-month period (Unaudited) | 8 | — | 8 | |||||||||
| Interest payable (Unaudited) | (4 | ) | — | (4 | ) | |||||||
| February 28, 2017 (Unaudited) | 1,389 | 309 | 1,698 | |||||||||
| Effective interest for the one-month period (Unaudited) | 31 | — | 31 | |||||||||
| Interest payable (Unaudited) | (14 | ) | — | (14 | ) | |||||||
| Effective interest for the thirteen-month period | 39 | — | 39 | |||||||||
| Interest payable during the period | (18 | ) | — | (18 | ) | |||||||
| March 31, 2017 | 1,406 | 309 | 1,715 | |||||||||
| Effective interest for the twelve-month period | 366 | — | 366 | |||||||||
| Interest payable during the period | (160 | ) | — | (160 | ) | |||||||
| March 31, 2018 | 1,612 | 309 | 1,921 |
| 13. | Capital and other components of equity |
| (a) | Share capital: |
Authorized capital stock:
Unlimited number of shares:
| Ø | Class A shares (Common Shares), voting (one vote per share), participating and without par value |
| Ø | Class B shares, voting (ten votes per share), non-participating, without par value and maximum annual non-cumulative dividend of 5% on the amount paid for said shares. Class B shares are convertible, at the holder’s discretion, into Class A shares (Common Shares), on a one-for-one basis, and Class B shares are redeemable at the holder’s discretion for $0.80 per share, subject to certain conditions. (1) |
| Ø | Class C shares, non-voting, non-participating, without par value and maximum annual non-cumulative dividend of 5% on the amount paid for said shares. Class C shares are convertible, at the holder’s discretion, into Class A shares (Common Shares), on a one-for-one basis, and Class C shares are redeemable at the holder’s discretion for $0.20 per share, subject to certain conditions. (1) |
| Ø | Class D and E shares, non-voting, non-participating, without par value and maximum monthly non-cumulative dividend between 0.5% and 2% on the amount paid for said shares. Class D and E shares are convertible, at the holder’s discretion, into Class A shares (Common Shares), on a one-for-one basis, and Class D and E shares are redeemable at the holder’s discretion, subject to certain conditions. (1) |
(1) None issued and outstanding
| F-29 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 13. | Capital and other components of equity (continued): |
| (b) | Public offering – December 27, 2017: |
On December 27, 2017, the Corporation closed a public offering issuing 9,900,990 units of Acasti (“Units”) at a price of US$1.01 per Unit for gross proceeds of $12.6 million (US$10 million). The units issued consist of 9,900,990 Class A shares (Common Shares) and 8,910,891 warrants with the right to purchase one Common Share (“Warrant”) of Acasti. As part of this closing, the underwriters’ also partially exercised for nil consideration the over-allotment option for warrants, which were issued for a right to purchase 892,044 Class A Common Shares at an exercise price of US$1.26.
On January 22, 2018, the underwritters exercised a portion of their over-allotment option by purchasing an additional 766,179 common shares at a price of US$1.01 per share, for additional gross proceeds of $963 (US$773).
The Warrants forming part of the Units are derivative liabilities (“Derivative Warrant Liabilities”) for accounting purposes due to the currency of the exercise price being different from the Corporation’s functional currency. The proceeds of the offering are required to be split between the Derivative Warrant Liabilities and the equity-classified Class A share at the time of issuance of the Units. The fair value of the Derivative Warrant Liabilities at the time of issuance was determined to be $5.9 million and the residual of the proceeds were allocated to the Class A shares. Total issue costs related to this transaction totaled approximately $2.7 million. The issue costs have been allocated between the Warrants and Class A shares based on relative value. The portion allocated to the Warrants was recognized in finance costs in the Statements of Earnings and Comprehensive Loss, whereas the portion allocated to Class A shares was recognized as a reduction to share capital, in the Statements of Financial Position.
The fair value of the public offering warrants in 2017 was estimated according to the Black-Scholes option pricing model and based on the following assumptions:
| December 27, 2017 |
||||
| Exercise price | US $1.26 | |||
| Share price | US $0.97 | |||
| Risk-free interest | 2.22 | % | ||
| Estimated life (in years) | 5 | |||
| Expected volatility | 93.52 | % |
The fair value of the public offering warrants issued was determined to be $0.60 per warrant as at December 27, 2017. Changes in the fair value of the Warrants are recognized in finance expenses.
As part of the transaction, the Company also issued broker warrants to purchase up to 495,050 Common Shares. Each Broker Warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of US$1.2625, at any time until December 27, 2022. The broker warrants are considered for compensation to non-employees under IFRS 2, stock-based compensation, and are accounted for at fair value at issuance date and not subsequently revalued. To determine the fair value of the Broker Warrants, the Black-Scholes pricing model was used based on the following assumptions:
| December 27, 2017 |
||||
| Exercise price | US $1.2625 | |||
| Share price | US $0.97 | |||
| Risk-free interest | 2.22 | % | ||
| Estimated life (in years) | 5 | |||
| Expected volatility | 93.52 | % |
The total cost associated with the Broker Warrants amounted to $406 and was allocated to contributed surplus.
| F-30 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 13. | Capital and other components of equity (continued): |
| (c) | Public offering - February 21, 2017: |
Concurrent with the private placement described in Note 12, on February 21, 2017, the Corporation closed a public offering (“Public Offering”) issuing 3,930,518 units of Acasti (“Units”) at a price of $1.45 per Unit for gross proceeds of $5,699. Each Unit consists of one class A share (Common Share) and one half of one class A or common share purchase warrant. Each whole warrant entitles the holder thereof to purchase one common share at an exercise price of $2.15 per common share, at any time until February 21, 2022. The Units issued as part of the public offering are considered equity instruments. The transaction costs associated with the Public Offering amounted to $1,190. The proceeds and transaction costs were allocated to share capital.
As part of the transaction, the Company also issued broker warrants (the “Broker Warrants”) to purchase up to 234,992 Common Shares. Each Broker Warrant entitles the holder thereof to acquire one Common Share of the Corporation at an exercise price of $2.15 per common share, at any time until February 21, 2018. The broker warrants are considered for compensation to non-employees under IFRS 2, stock-based compensation, and are accounted for at fair value through contributed surplus. To determine the fair value of the Broker Warrants, the Black-Scholes pricing model was used. The total costs associated with the Broker Warrants amounted to $144 and were allocated to share capital.
The warrants issued as part of the Units of the Public Offering and the broker warrants include an “Acceleration Right”, related to the Corporation’s right to accelerate the expiry date of the warrants. The Acceleration Right clause means the right of the Corporation to accelerate the expiry date to a date that is not less than 30 days following delivery of the acceleration notice if, at any time at least four months after the effective date, the volume weighted average trading price of the common shares equals or exceeds $2.65 for a period of 20 consecutive trading days on the TSXV.
Furthermore, as part of the February 2017 Public Offering and convertible debt transactions, a total of 60,000 Common Shares were issued as equity settled share-based payments for services received from an employee of the previous parent at a price of $1.57 per share for a total cost of $94. The equity settled share-based payment costs have been allocated to share capital for a cost that amounted to $85 and to debt for a cost that amounted to $9 based on relative value.
The value of the broker warrants was estimated using the Black-Scholes option pricing model and based on the following assumptions:
| February 21, 2017 | ||||
| Exercise price | $2.15 | |||
| Share price | $1.70 | |||
| Dividend | — | |||
| Risk-free interest | 0.79 | % | ||
| Estimated life (in years) | 1.00 | |||
| Expected volatility | 112.09 | % |
The total cost associated with the Broker Warrants amounted to $144 and was allocated to contributed surplus.
| (d) | Issuance of shares: |
The following table summarizes the shares issued to settle the payment of accrued interest on the unsecured convertible debentures with the corresponding amount recorded to share capital.
| Accrued interest as at | Share issuance date | Number of shares | Amount $ |
|||||||||
| March 31, 2017 | April 7, 2017 | 9,496 | 17 | |||||||||
| June 30, 2017 | August 15, 2017 | 23,885 | 40 | |||||||||
| September 30, 2017 | December 27, 2017 | 22,783 | 40 | |||||||||
| December 31, 2017 | March 27, 2018 | 33,605 | 40 | |||||||||
| 89,769 | 137 |
| F-31 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 13. | Capital and other components of equity (continued): |
| (e) | Warrants: |
The warrants of the Corporation are composed of the following as at March 31, 2018, March 31, 2017 and February 28, 2017:
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
February 29, 2016 | |||||||||||||||||||||||||||||
| Number outstanding |
Amount | Number outstanding |
Amount | Number outstanding | Amount | Number outstanding | Amount | |||||||||||||||||||||||||
| $ | $ | $ | $ | |||||||||||||||||||||||||||||
| Liability | ||||||||||||||||||||||||||||||||
| Series December 2017 | ||||||||||||||||||||||||||||||||
| US public offering Warrants 2017 (i) | 9,802,935 | 6,405 | — | — | — | — | — | — | ||||||||||||||||||||||||
| Series 8 Public offering | ||||||||||||||||||||||||||||||||
| Warrants December 2013 (note 11) (ii) | 18,400,000 | 21 | 18,400,000 | 209 | 18,400,000 | 187 | 18,400,000 | 156 | ||||||||||||||||||||||||
| 28,202,935 | 6,426 | 18,400,000 | 209 | 18,400,000 | 187 | 18,400,000 | 156 | |||||||||||||||||||||||||
| Equity | ||||||||||||||||||||||||||||||||
| Public offering warrants | ||||||||||||||||||||||||||||||||
| Series December 2017 US Broker warrants (v) | 495,050 | 406 | — | — | — | — | — | — | ||||||||||||||||||||||||
| Series 2017 BW Broker warrants (iii) | — | — | 234,992 | 144 | 234,992 | 144 | — | — | ||||||||||||||||||||||||
| Public offering warrants February 2017 (iv) | 1,904,034 | — | 1,965,259 | — | 1,965,259 | — | — | — | ||||||||||||||||||||||||
| Private Placement – contingent warrants | ||||||||||||||||||||||||||||||||
| 2017 Unsecured convertible debenture conversion option and contingent warrants (vi) | 1,052,630 | 309 | 1,052,630 | 309 | 1,052,630 | 309 | — | — | ||||||||||||||||||||||||
| Series 9 Private Placement warrants 2013 (vii) | 161,654 | — | 161,654 | — | 161,654 | — | 161,654 | — | ||||||||||||||||||||||||
| 3,613,368 | 715 | 3,414,535 | 453 | 3,414,535 | 453 | 161,654 | — | |||||||||||||||||||||||||
| (i) | Warrant to acquire one Common Share of the Corporation at an exercise price of US$1.26, expiring on December 27, 2022. |
| (ii) | In order to obtain one Common Share of the Corporation at an exercise price of US$15.00, 10 warrants must be exercised. Warrants expire on December 3, 2018. |
| (iii) | Warrant to acquire one Common Share of the Corporation at an excersise price of 2.15 expiring on February 21, 2018. 117,496 warrants amounted to $71 were exercised in November 2017 and 117,496 warrants expired on February 21, 2018. |
| (iv) | Warrant to acquire one Common Share of the Corporation at an exercise price of US$1.2625, expiring on December 27, 2022. 61,225 warrants amounted to $132 were exercised in November 2017. |
| (v) | Warrant to acquire one Common Share of the Corporation at an exercise price of $2.15, expiring on February 21, 2022. |
| (vi) | Warrant to acquire one Common Share of the Corporation at an exercise price of $1.90 expiring on February 21, 2020, net of deferred tax expense of $129. |
| (vii) | Warrant to acquire one Common Share of the Corporation at an exercise price of $13.30, expiring on December 3, 2018. |
| F-32 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 14. | Personnel expenses: |
| Thirteen- months ended |
Month ended | Twelve-month period ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Salaries and other short-term employee benefits | 3,281 | 2,491 | 214 | 2,277 | 1,902 | |||||||||||||||
| Share-based compensation costs | 929 | 674 | 86 | 588 | 309 | |||||||||||||||
| Severance | — | — | — | — | 210 | |||||||||||||||
| Total personnel expenses | 4,210 |
3,165 |
300 |
2,865 |
2,421 |
| 15. | Financial expenses: |
| Thirteen- months ended |
Month ended | Twelve-month period ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Interest income | 72 | 125 | 6 | 119 | 73 | |||||||||||||||
| Foreign exchange gain | - | - | - | - | 1,023 | |||||||||||||||
| Financial income | 72 | 125 | 6 | 119 | 1,096 | |||||||||||||||
| Foreign exchange loss | (32 | ) | (180 | ) | (3 | ) | (177 | ) | - | |||||||||||
| Interest payable on convertible debenture during the period | (160 | ) | (17 | ) | (14 | ) | (3 | ) | - | |||||||||||
| Accretion of interest on convertible debenture | (206 | ) | (22 | ) | (17 | ) | (5 | ) | - | |||||||||||
| Transaction costs related to derivative warrant liabilities | (1,134 | ) | - | - | - | - | ||||||||||||||
| Other charges | (4 | ) | (19 | ) | (1 | ) | (18 | ) | (2 | ) | ||||||||||
| Financial expenses | (1,536 | ) | (238 | ) | (35 | ) | (203 | ) | (2 | ) | ||||||||||
| Other net financial expenses | (1,464 | ) | (113 | ) | (29 | ) | (84 | ) | 1,094 | |||||||||||
| Change in fair value of warrant liabilities | (344 | ) | (53 | ) | (22 | ) | (31 | ) | 2,201 | |||||||||||
| Net Financial expenses | (1,808 | ) | (166 | ) | (51 | ) | (115 | ) | 3,295 |
| F-33 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 16. | Share-based payments: |
At March 31, 2018, the Corporation has the following share-based payment arrangement:
| (a) | Corporation stock option plan: |
The Corporation has in place a stock option plan for directors, officers, employees and consultants of the Corporation. The plan provides for the granting of options to purchase Class A shares (Common Shares). The exercise price of the stock options granted under this plan is not lower than the closing price of the shares listed on the TSXV at the close of markets the day preceding the grant. Under this plan, the maximum number of Class A shares (Common Shares) that may be issued upon exercise of options granted under the plan is 2,940,511, representing 20% of the number of Class A shares (Common Shares) issued and outstanding as at February 29, 2016. The terms and conditions for acquiring and exercising options are set by the Corporation’s Board of Directors, subject among others, to the following limitations: the term of the options cannot exceed ten years and every stock option granted under the stock option plan will be subject to conditions no less restrictive than a minimum vesting period of 18 months and a gradual and equal acquisition of vesting rights not shorter than on a quarterly basis. The total number of shares issued to any one consultant cannot exceed 2% of the Corporation’s total issued and outstanding shares. The Corporation is not authorized to grant such number of options under the stock option plan that could result in a number of Class A shares (Common Shares) issuable pursuant to options granted to (a) related persons exceeding 10% of the Corporation’s issued and outstanding Class A shares (Common Shares) (on a non-diluted basis) on the date an option is granted, or (b) any one eligible person in a twelve month period exceeding 5% of the Corporation’s issued and outstanding Class A shares (Common Shares) (on a non-diluted basis) on the date an option is granted.
The following tables summarize information about activities within the stock option plan:
| March 31, 2018 | Thirteen-month period ended March 31, 2017 |
|||||||||||||||
| Weighted average exercise price |
Number of options |
Weighted average exercise price |
Number of options |
|||||||||||||
| $ | $ | |||||||||||||||
| Outstanding at beginning of period | 2.58 | 1,424,788 | 13.52 | 454,151 | ||||||||||||
| Granted | 1.75 | 1,121,500 | 1.69 | 1,300,400 | ||||||||||||
| Forfeited | 1.89 | (199,800 | ) | 13.27 | (190,138 | ) | ||||||||||
| Expired | 18.06 | (62,100 | ) | 15.38 | (139,625 | ) | ||||||||||
| Outstanding at end of period | 1.81 | 2,284,388 | 2.58 | 1,424,788 | ||||||||||||
| Exercisable at end of period | 1.92 | 591,113 | 6.44 | 238,482 | ||||||||||||
| Month ended March 31, 2017 |
Twelve-month period ended February 28, 2017 |
|||||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Weighted average exercise price |
Number of options |
Weighted average exercise price |
Number of options |
|||||||||||||
| $ | $ | |||||||||||||||
| Outstanding at beginning of period | 2.59 | 1,427,288 | 13.52 | 454,151 | ||||||||||||
| Granted | — | — | 1.69 | 1,300,400 | ||||||||||||
| Forfeited | 11.50 | (2,500 | ) | 13.29 | (187,638 | ) | ||||||||||
| Expired | — | — | 15.38 | (139,625 | ) | |||||||||||
| Outstanding at end of period | 2.58 | 1,424,788 | 2.59 | 1,427,288 | ||||||||||||
| Exercisable at end of period | 6.44 | 238,482 | 6.49 | 240,982 | ||||||||||||
| F-34 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 16. | Share-based payments (continued): |
| (a) | Corporation stock option plan (continued): |
| February 29, 2016 | ||||||||
| Weighted average exercise price |
Number of options |
|||||||
| $ | ||||||||
| Outstanding at beginning of period | 15.33 | 429,625 | ||||||
| Granted | 4.65 | 109,188 | ||||||
| Exercised | 2.50 | (250 | ) | |||||
| Forfeited | 9.40 | (66,912 | ) | |||||
| Expired | 18.57 | (17,500 | ) | |||||
| Outstanding at end of period | 13.52 | 454,151 | ||||||
| Exercisable at end of period | 15.28 | 375,563 | ||||||
The weighted average of the fair value of the options granted to employees and directors of the Company during the period ended March 31, 2018 is $1.22 (thirteen-month period ended March 31, 2017 is $1.40 and during the twelve-month period ended February 28, 2017 is $1.40 (unaudited) (2016 - $2.14)). There were no options granted during the month ended March 31, 2017 and no options granted to consultants during the thirteen-month period ended March 31, 2017 and years ended February 29, 2016.
No options were exercised during the period ended March 31, 2018 (nil for the thirteen-month period ended March 31, 2017). The weighted average share price at the date of exercise for share options exercised during the year ended February 29, 2016 was $4.20. Stock-based compensation recognized under this plan for the period ended March 31, 2018 was $929 (thirteen-month and one-month periods ended March 31, 2017 amounted to $674 and $86 (unaudited), respectively and amounted to $588 (unaudited) for the twelve-month period ended February 28, 2017 and $234 for 2016).
The fair value of options granted was estimated using the Black-Scholes option pricing model, resulting in the following weighted average assumptions for options granted during the periods ended:
| Thirteen-month period ended |
Twelve-month Period ended |
|||||||||||||||
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
February 29, 2016 | |||||||||||||
| Exercise price | $1.75 | $1.69 | $1.69 | $4.65 | ||||||||||||
| Share price | $1.75 | $1.69 | $1.69 | $4.65 | ||||||||||||
| Dividend | — | — | — | — | ||||||||||||
| Risk-free interest | 1.21 | % | 0.87 | % | 0.87 | % | 0.66 | % | ||||||||
| Estimated life (in years) | 5.89 | 4.94 | 4.94 | 4.20 | ||||||||||||
| Expected volatility | 82.4 | % | 123.5 | % | 123.5 | % | 65.63 | % |
The expected life of the stock options is based on historical data and current expectation and is not necessarily indicative of exercise patterns that may occur. The expected volatility reflects the assumption that the historical volatility over a period similar to the life of the options is indicative of future trends, which may also not necessarily be the actual outcome.
| F-35 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 16. | Share-based payments (continued): |
| (a) | Corporation stock option plan (continued): |
The following tables summarize the status of the outstanding and exercisable options of the Corporation:
| March 31, 2018 | ||||||||||||||||||
| Options outstanding | Exercisable options | |||||||||||||||||
| Exercise price | Weighted
remaining contractual life outstanding |
Number
of options outstanding |
Weighted
average exercise price $ |
Number
of options exercisable |
||||||||||||||
| $1.56 | - | $1.58 | 5.11 | 525,000 | 1.56 | 306,250 | ||||||||||||
| $1.59 | - | $1.71 | 8.90 | 415,000 | 1.65 | 141,667 | ||||||||||||
| $1.72 | - | $1.88 | 9.20 | 992,500 | - | - | ||||||||||||
| $1.89 | - | $2.25 | 5.16 | 286,700 | 1.99 | 95,568 | ||||||||||||
| $2.26 | - | $6.50 | 3.67 | 65,188 | 4.87 | 47,628 | ||||||||||||
| 7.54 | 2,284,388 | 1.92 | 591,113 | |||||||||||||||
Share-based payment transactions and broker warrants:
The fair value of share-based payment transaction is measured using the Black-Scholes valuation model. Measurement inputs include share price on measurement date, exercise price of the instrument, expected volatility (based on weighted average historic volatility), weighted average expected life of the instruments (based on historical experience and general option holder behaviour unless no entity-specific information exists in which case the average of the vesting and contractual periods is used), expected dividends, and the risk-free interest rate (based on government bonds). Service and non-market performance conditions attached to the transactions, if any, are not taken into account in determining fair value.
| b) | Corporation equity incentive plan: |
The Corporation established an equity incentive plan for employees, directors and consultants. The plan provides for the issuance of restricted share units (“RSU”), performance share units, restricted shares, deferred share units and other share-based awards, subject to restricted conditions as may be determined by the Board of Directors. There are no such awards outstanding as of March 31, 2018, March 31, 2017, and February 28, 2017 and no stock-based compensation was recognized for the period ended March 31, 2018 (nil for the one-month and thirteen-month periods ended March 31, 2017 and $64 for the twelve-month period ended February 29, 2016).
| 17. | Loss per share: |
Diluted loss per share was the same amount as basic loss per share, as the effect of options, RSUs and warrants would have been anti-dilutive, because the Corporation incurred losses in each of the periods presented. All outstanding options, RSUs and warrants could potentially be dilutive in the future.
| F-36 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 18. | Supplemental cash flow disclosure: |
| (a) | Changes in working capital items: |
| Thirteen-months ended |
Month ended | Twelve-months ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Receivables | (553 | ) | 193 | (40 | ) | 233 | 406 | |||||||||||||
| Receivable from corporation under common control | - | - | - | - | 50 | |||||||||||||||
| Inventories | - | - | - | - | 88 | |||||||||||||||
| Prepaid expenses | (103 | ) | 247 | (33 | ) | 280 | (138 | ) | ||||||||||||
| Other Assets | (659 | ) | - | - | - | - | ||||||||||||||
| Trade and other payables | 4,898 | 352 | (255 | ) | 607 | (447 | ) | |||||||||||||
| Total changes in working capital items | 3,583 | 792 | (328 | ) | 1,120 | (41 | ) |
| F-37 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 18. | Supplemental cash flow disclosure (continued): |
| (b) | Non-cash transactions: |
| Thirteen-months ended |
Month ended |
Twelve-months ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Equity settled share-based payment included in equity | 137 | 94 | — | 94 | — | |||||||||||||||
| Issuance of broker warrants included in net proceeds from public offering | 406 | 144 | — | 144 | — | |||||||||||||||
| Public offering transaction costs included in trade and other payables | 132 | 381 | 381 | 416 | — | |||||||||||||||
| Reduction in share issue costs from reduction in trade and other payables | — | 109 | — | 109 | — | |||||||||||||||
| Private Placement transaction costs included in trade and other payables | — | 40 | 40 | 50 | — | |||||||||||||||
| Equipment included in trade and other payables | 216 | 288 | 288 | 269 | — | |||||||||||||||
| Interest payable included in trade and other payables | 40 | 18 | 18 | 4 | — | |||||||||||||||
| Issuance of shares on settlement of a liability | — | — | — | — | 103 | |||||||||||||||
| Interest receivable included in payable to Neptune corporation | — | — | — | — | 27 |
| F-38 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 19. | Income taxes: |
Deferred tax (recovery) expense:
| Thirteen-months ended | Month ended |
Twelve-months ended |
||||||||||||||||||
| March 31, 2018 |
March 31, 2017 |
March 31, 2017 |
February 28, 2017 |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Origination and reversal of temporary differences | 5,241 | 2,240 | 163 | 2,077 | 2,065 | |||||||||||||||
| Change in unrecognized deductible temporary differences | (5,241 | ) | (2,369 | ) | (163 | ) | (2,206 | ) | (2,065 | ) | ||||||||||
| Deferred tax (recovery) expense | — | (129 | ) | — | (129 | ) | — |
Reconciliation of effective tax rate:
|
Thirteen-months ended |
Month ended |
Twelve-months ended |
||||||||||||||||||
|
March 31, 2018 |
March 31, 2017 |
March 31, (Unaudited) |
February 28, 2017 (Unaudited) |
February 29, 2016 |
||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
| Loss before income taxes |
(21,504 |
) |
(11,376 |
) | (769 | ) |
(10,607 |
) |
(6,317 |
) | ||||||||||
Basic combined Canadian statutory income tax rate 1 |
26.78 | % | 26.87 | % | 26.80 | % | 26.88 | % | 26.90 | % | ||||||||||
| Computed income tax recovery | (5,759 | ) | (3,057 | ) | (206 | ) | (2,851 | ) | (1,699 | ) | ||||||||||
| Increase resulting from: | ||||||||||||||||||||
| Change in unrecognized deductible temporary differences | 5,241 | 2,369 | 162 | 2,207 | 2,065 | |||||||||||||||
| Non-deductible stock-based compensation | 248 | 178 | 23 | 155 | 83 | |||||||||||||||
| Non-deductible change in fair value | 92 | 14 | 6 | 8 | (592 | ) | ||||||||||||||
| Permanent differences and other | 118 | 166 | 12 | 154 | 143 | |||||||||||||||
| Change in statutory income tax rate | 60 | 201 | 3 | 198 | — | |||||||||||||||
| Total tax (recovery) expense | — | (129 | ) | — | (129 | ) | — | |||||||||||||
1 The Canadian combined statutory income tax rate has decreased due to a reduction in the provincial statutory income tax rate.
| F-39 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 19. | Income taxes (continued): |
Unrecognized deferred tax assets:
At March 31, 2018, March 31, 2017 and February 28, 2017, the net deferred tax assets, which have not been recognized in these financial statements because the criteria for recognition of these assets were not met, were as follows:
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||
| $ | $ | $ | ||||||||||
| Deferred tax assets | ||||||||||||
| Tax losses carried forward | 12,670 | 8,293 | 8,153 | |||||||||
| Research and development expenses | 4,927 | 4,220 | 4,196 | |||||||||
| Property, plan and equipment and intangible assets | 567 | 435 | 423 | |||||||||
| Other deductible temporary differences | 884 | 522 | 539 | |||||||||
| Deferred tax assets | 19,048 | 13,470 | 13,311 | |||||||||
| Deferred tax liabilities | ||||||||||||
| Tax basis of unsecured convertible debentures in excess of carrying value | 67 | 122 | 126 | |||||||||
| Deferred tax liabilities | 67 | 122 | 126 | |||||||||
| Net deferred tax assets | 18,981 | 13,348 | 13,185 |
On initial recognition of the unsecured convertible debenture equity component on February 21, 2017, a deferred tax liability of $129 was recognized with the corresponding entry recognized directly in Other equity. Consequently, an equal amount of deferred tax asset related to unrecognized tax losses was recognized with the offsetting entry in the Corporation statement of earnings and comprehensive loss.
As at March 31, 2018, the amounts and expiry dates of tax attributes and temporary differences, which are available to reduce future years’ taxable income, were as follows:
| March 31, 2018 | ||||||||
| Federal | Provincial | |||||||
| $ | $ | |||||||
| Tax losses carried forward | ||||||||
| 2029 | 714 | 714 | ||||||
| 2030 | 1,627 | 1,620 | ||||||
| 2031 | 2,071 | 2,063 | ||||||
| 2032 | 2,262 | 2,241 | ||||||
| 2033 | 1,854 | 1,825 | ||||||
| 2034 | 3,598 | 3,598 | ||||||
| 2035 | 4,595 | 4,459 | ||||||
| 2036 | 5,494 | 5,494 | ||||||
| 2037 | 8,584 | 8,456 | ||||||
| 2038 | 17,155 | 17,155 | ||||||
| 47,954 | 47,625 | |||||||
| Research and development expenses, without time limitation | 18,002 | 19,362 | ||||||
| Other deductible temporary differences, without time limitation | 5,224 | 5,224 | ||||||
| F-40 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 20. | Financial instruments: |
This note provides disclosures relating to the nature and extent of the Corporation’s exposure to risks arising from financial instruments, including credit risk, foreign currency risk, interest rate risk and liquidity risk, and how the Corporation manages those risks.
| (a) | Credit risk: |
Credit risk is the risk of a loss if a customer or counterparty to a financial asset fails to meet its contractual obligations. The Corporation has credit risk relating to cash and cash equivalents and short-term investments, which it manages by dealing only with highly-rated Canadian institutions. The carrying amount of financial assets, as disclosed in the statements of financial position, represents the Corporation’s credit exposure at the reporting date.
| (b) | Currency risk: |
The Corporation is exposed to the financial risk related to the fluctuation of foreign exchange rates and the degrees of volatility of those rates. Foreign currency risk is limited to the portion of the Corporation's business transactions denominated in currencies other than the Canadian dollar. Fluctuations related to foreign exchange rates could cause unforeseen fluctuations in the Corporation's operating results.
A portion of the expenses, mainly related to research contracts and purchase of production equipment, is incurred in US dollars and in Euros, for which no financial hedging is required. There is a financial risk related to the fluctuation in the value of the US dollar and the Euro in relation to the Canadian dollar. In order to minimize the financial risk related to the fluctuation in the value of the US dollar in relation to the Canadian dollar, funds continue to be invested as short-term investments in the US dollar.
The following table provides an indication of the Corporation’s significant foreign exchange currency exposures as stated in Canadian dollars at the following dates:
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||||||||||||||
| US $ |
Euro | US $ |
Euro | US $ |
Euro | |||||||||||||||||||
| Cash and cash equivalents | 7,024 | — | 3,524 | — | 3,691 | — | ||||||||||||||||||
| Marketable securities | 26 | — | — | — | — | — | ||||||||||||||||||
| Receivables | 6 | — | 2 | — | 3 | — | ||||||||||||||||||
| Trade and other payables | (3,924 | ) | (627 | ) | (503 | ) | (317 | ) | (376 | ) | (603 | ) | ||||||||||||
| 3,132 | (627 | ) | 3,023 | (317 | ) | 3,318 | (603 | ) | ||||||||||||||||
The following exchange rates are those applicable to the following periods and dates:
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||||||||||||||
| Average | Reporting | Average | Reporting | Average | Reporting | |||||||||||||||||||
| CA$ per US$ | 1.2834 | 1.2900 | 1.3134 | 1.3299 | 1.3113 | 1.3281 | ||||||||||||||||||
| CA$ per Euro | 1.5008 | 1.5898 | 1.4424 | 1.4251 | 1.4434 | 1.4066 | ||||||||||||||||||
| F-41 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 20. | Financial instruments (continued): |
| (b) | Currency risk (continued): |
Based on the Corporation’s foreign currency exposures noted above, varying the above foreign exchange rates to reflect a 5% strengthening of the US dollar and Euro would have decrease in net loss as follows, assuming that all other variables remain constant:
| March 31, 2018 | March 31, 2017 | February 29, 2017 (Unaudited) |
||||||||||
| $ | $ | $ | ||||||||||
| Decrease in net loss | 88 | 139 | 151 |
An assumed 5% weakening of the foreign currencies would have an equal but opposite effect on the basis that all other variables remained constant.
| (c) | Interest rate risk: |
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market rates.
The Corporation’s exposure to interest rate risk as at March 31, 2018, March 31, 2017and February 28, 2017 is as follows:
| Cash and cash equivalents | Short-term fixed interest rate | |
| Unsecured convertible debentures | Long-term fixed interest rate |
The capacity of the Corporation to reinvest the short-term amounts with equivalent return will be impacted by variations in short-term fixed interest rates available on the market. Management believes that the risk the Corporation will realize a loss as a result of the decline in the fair value of its cash equivalents is limited because these investments have short-term maturities and are generally held to maturity.
| (d) | Liquidity risk: |
Liquidity risk is the risk that the Corporation will not be able to meet its financial obligations as they fall due. The Corporation manages liquidity risk through the management of its capital structure and financial leverage, as outlined in Note 22. It also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves the Corporation's operating budgets, and reviews material transactions outside the normal course of business. Refer to Note 2(c).
| F-42 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 20. | Financial instruments (continued): |
| (d) | Liquidity risk (continued): |
The following are the contractual maturities of financial liabilities as at March 31, 2018, March 31, 2017and February 28, 2017:
| March 31, 2018 | ||||||||||||||||||
| Required payments per year | Total | Carrying amount | Less than 1 year | 1 to 3 years | ||||||||||||||
| Notes | $ | $ | $ | $ | ||||||||||||||
| Trade and other payables | 10 | 6,697 | 6,697 | 6,697 | — | |||||||||||||
| Unsecured convertible debentures |
12 |
2,303 | 1,612 | 160 | 2,143 | |||||||||||||
| 9,000 | 8,309 | 6,857 | 2,143 |
| March 31, 2017 | ||||||||||||||||||
| Required payments per year | Total | Carrying amount | Less than 1 year | 1 to 3 years | ||||||||||||||
| Notes | $ | $ | $ | $ | ||||||||||||||
| Trade and other payables | 10 | 2,138 | 2,138 | 2,138 | — | |||||||||||||
| Unsecured convertible debentures | 12 | 2,463 | 1,406 | 160 | 2,303 | |||||||||||||
| 4,601 | 3,544 | 2,298 | 2,303 |
| February 28, 2017 (Unaudited) |
||||||||||||||||||
| Required payments per year | Total | Carrying amount | Less than 1 year | 1 to 3 years | ||||||||||||||
| Notes | $ | $ | $ | $ | ||||||||||||||
| Trade and other payables | 10 | 2,405 | 2,405 | 2,405 | — | |||||||||||||
| Unsecured convertible debentures | 12 | 2,476 | 1,389 | 160 | 2,316 | |||||||||||||
| 4,881 | 3,794 | 2,565 | 2,316 |
The Derivative warrant liabilities are excluded from the above tables as they will be settled in shares and not by the use of liquidities.
| 21. | Commitments and contingencies: |
Research and development contracts and contract research organizations agreements:
The Company utilizes contract manufacturing organizations related to the development of clinical material and clinical research organizations to perform services related to the Company’s clinical trials. Pursuant to these agreements with manufacturing and contract research organizations, the Company has the right to terminate the agreements either without penalties or under certain penalty conditions. For agreements which contain penalty conditions, the Company would be required to pay penalties of approximately $172.
During the year, the Company entered into a lease agreement, for its research and development and quality control laboratory facility located in Sherbrooke, Québec, resulting in a total commitment of $151 over the two-year lease term. An amount of $72 is committed in the next year, with a remaining committed amount of $79 over the second year of the lease.
Contingencies:
A former CEO of the Corporation is claiming the payment of approximately $8.5 million and the issuance of equity instruments from the Group. As the Corporation’s management believes that these claims are not valid, no provision has been recognized. Neptune and its subsidiaries also filed an additional claim to recover certain amounts from the former officer. All outstanding share-based payments held by the former CEO have been cancelled during the year ended February 28, 2015.
| F-43 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 21. | Commitments and contingencies (continued): |
The Corporation is also involved in other matters arising in the ordinary course of its business. Since management believes that all related claims are not valid and it is presently not possible to determine the outcome of these matters, no provisions have been made in the financial statements for their ultimate resolution beyond the amounts incurred and recorded for such matters. The resolution of such matters could have an effect on the Corporation’s financial statements in the year that a determination is made, however, in management’s opinion, the final resolution of all such matters is not projected to have a material adverse effect on the Corporation’s financial position.
| 22. | Determination of fair values: |
Certain of the Corporation’s accounting policies and disclosures require the determination of fair value, for both financial assets and liabilities. Fair values have been determined for measurement and/or disclosure purposes based on the following methods.
Financial assets and liabilities:
In establishing fair value, the Corporation uses a fair value hierarchy based on levels as defined below:
| · | Level 1: defined as observable inputs such as quoted prices in active markets. |
| · | Level 2: defined as inputs other than quoted prices in active markets that are either directly or indirectly observable. |
| · | Level 3: defined as inputs that are based on little or no observable market data, therefore requiring entities to develop their own assumptions. |
The Corporation has determined that the carrying values of its short-term financial assets and liabilities approximate their fair value given the short-term nature of these instruments. The fair value of the liability component of the convertible debenture is determined by discounting future cash flows using a rate that the Corporation could obtain for loans with similar terms, conditions and maturity dates. The fair value of this liability at March 31, 2018 approximates the carrying amount and was measured using level 3 inputs.
Derivative warrant liabilities:
The Corporation measured its derivative warrant liabilities at fair value on a recurring basis. These financial liabilities were measured using a level 3 inputs (Note 11).
As at March 31, 2018, the effect of an increase or a decrease of 5% of the volatility used, which is the significant unobservable input in the fair value estimate, would result in a loss of $241 or a gain of $254, respectively.
As at March 31, 2018, the effect of a 5% strengthening of the US dollar, would result in a loss of $320. An assumed 5% weakening of the foreign currency would have an equal but opposite effect on the basis that all other variables remained constant.
| 23. | Capital management: |
Since inception, the Corporation’s objective in managing capital is to ensure sufficient liquidity to finance its research and development activities, general and administrative expenses, expenses associated with intellectual property protection and its overall capital expenditures. The Corporation is not exposed to external requirements by regulatory agencies or third parties regarding its capital, except for certain covenants included within the convertible debentures (Note 12).
Since the beginning of its operations, the Corporation has primarily financed its liquidity needs from funding provided through public offerings, private placements, from the exercise of warrants that were distributed to its related party’s shareholders, from a rights offering and from the issuance of options to employees.
The Corporation defines capital to include total shareholders’ equity, derivative warrant liabilities and unsecured convertible debentures.
The Corporation’s policy is to maintain a minimal level of debt.
| F-44 |
| ACASTI PHARMA INC. |
| Notes to Financial Statements |
| For the year ended March 31, 2018 and the thirteen-month and one-month periods ended March 31, 2017, and the twelve-month period ended February 28, 2017 year ended February 29, 2016 |
| (thousands of Canadian dollars, except where noted and for share and per share amounts) |
| 23. | Capital management (continued): |
The following table summarizes the cash and cash equivalents of the Corporation:
| March 31, 2018 | March 31, 2017 | February 28, 2017 (Unaudited) |
||||||||||
| Cash | 1,583 | 6,778 | 7,584 | |||||||||
| Cash equivalents | 6,640 | 2,994 | 2,989 | |||||||||
| Total Cash and cash equivalents | 8,223 | 9,772 | 10,573 |
As at March 31, 2018, cash equivalents consist of four term deposits totaling $4,193 (US - $3,250), two commercial paper totaling $1,418 (US - $1,099) and one promissory note totaling $ 1,029 (US- $798), each being held with a Canadian financial institution having a high credit rating. The term deposits, commercial paper and promissory note have maturity dates of ranging between April 2, 2018 and May 11, 2018, bearing interest rates ranging from 1.26% and 1.72% per annum, cashable at any time at the discretion of the Corporation, under certain conditions.
As at March 31, 2018, the Corporation held a marketable security of a term deposit totaling $26 (US - $20) held as restricted with maturity of March 13, 2019 and bearing interest at 2.23%.
As at March 31, 2017 and February 28, 2017, cash equivalents consisting of two term deposits totaling $2,994 (US - $2,251) and $2,990 (US$2,251) (unaudited), respectively, are being held with a Canadian financial institution having a high credit rating. The term deposits as at March 31, 2017 have maturity dates of April 11, 2017 and April 25, 2017, bearing an interest rate of 0.52% and 0.53% per annum, respectively, cashable at any time at the discretion of the Corporation, under certain conditions. The term deposits as at February 28, 2017 have maturity dates of March 12, 2017 and March 28, 2017, bearing an interest rate of 0.46% and 0.45% per annum, respectively, cashable at any time at the discretion of the Corporation, under certain conditions.
| 24. | Subsequent event |
On May 9, 2018, the Company announced the closing of a Canadian public offering of Units of the Company at a price of CA$1.05 per Unit for aggregate gross proceeds of approximately CA$10 million generating net proceeds of approximately CA$8.7 million. The Company issued 9,530,000 Units. Each Unit is comprised of one common share and one common share purchase warrant of the Company, exercisable at any time prior to May 9, 2023 at an exercise price of CA$1.31 per common share.
On May 14, 2018, the Company announced the full exercise of the over-allotment option for additional gross proceeds of approximately $1.5 million generating net proceeds to the Company of approximately CA$1.3 million. Pursuant to the over-allotment option, the Company issued an additional aggregate of 1,429,500 Units at the CA$1.05 offering price. Each Unit is also comprised of one common share and one common share purchase warrant of the Company exercisable at any time prior to May 9, 2023 at an exercise price of CA$1.31 per Common Share.
As consideration of services rendered by the Underwriter in connection with this offering and its over-allotment exercise, the Company paid the Underwriter a cash commission equal to 7% of the gross proceeds raised under the offering and granted the Underwriter non-transferable broker warrants equal to 5% at an exercise price equal to the CA$1.05 offering price. A Total of 547,975 broker warrants were issued to the Underwriters to purchase up to 547,975 common share of the Corporation at an exercise price of CA$1.05. 476,500 broker warrants will expire on May 9, 2023 and 71,475 will expire on May 14, 2023.
F-45